Last Update: 7 Dec 2018
RMarkdown:
generate-boxplots.Rmd
Using R to create boxplots
For datasets that have many observations per sample and you want to visualize their distribution, boxplots are good choice. Here, the example dataset has average values of multiple observations for each sample.
1
wget https://bioinformaticsworkbook.org/tutorials/data/mythical_creatures.txt
It is organized in this way:
Table 1: Gene length for 4 different gene families and for 3 different organism.
| Organism | gene_family_1 | gene_family_2 | gene_family_3 | gene_family_4 |
|---|---|---|---|---|
| Unicorn | 263 | 407 | 460 | 519 |
| Unicorn | 269 | 644 | 409 | 904 |
| Unicorn | 705 | 592 | 489 | 831 |
| Unicorn | 501 | 368 | 245 | 1269 |
| Unicorn | 244 | 520 | 403 | 227 |
| Unicorn | 387 | 275 | 280 | 185 |
| Unicorn | 437 | 93 | 391 | 353 |
| Unicorn | 552 | 612 | 476 | 1289 |
| Unicorn | 745 | 593 | 347 | 1453 |
| Unicorn | 751 | 507 | 155 | 820 |
| Cyclops | 583 | 463 | 153 | 1303 |
| Cyclops | 769 | 329 | 211 | 274 |
| Cyclops | 459 | 452 | 283 | 1289 |
| Cyclops | 149 | 568 | 210 | 280 |
| Cyclops | 181 | 314 | 412 | 1333 |
| Cyclops | 693 | 176 | 359 | 361 |
| Cyclops | 208 | 561 | 161 | 842 |
| Cyclops | 202 | 365 | 489 | 259 |
| Cyclops | 336 | 371 | 347 | 168 |
| Cyclops | 334 | 548 | 358 | 419 |
| Midwestern Dragon | 404 | 76 | 434 | 1188 |
| Midwestern Dragon | 325 | 466 | 259 | 1436 |
| Midwestern Dragon | 713 | 218 | 277 | 837 |
| Midwestern Dragon | 485 | 297 | 346 | 1258 |
| Midwestern Dragon | 652 | 527 | 325 | 750 |
| Midwestern Dragon | 632 | 342 | 309 | 1352 |
| Midwestern Dragon | 800 | 581 | 497 | 870 |
| Midwestern Dragon | 623 | 323 | 354 | 206 |
| Midwestern Dragon | 733 | 123 | 244 | 646 |
| Midwestern Dragon | 751 | 477 | 474 | 1390 |
| Sun Drop Flower | 743 | 100 | 473 | 625 |
| Sun Drop Flower | 160 | 230 | 164 | 1207 |
| Sun Drop Flower | 260 | 569 | 493 | 650 |
| Sun Drop Flower | 443 | 156 | 340 | 775 |
| Sun Drop Flower | 366 | 380 | 312 | 167 |
| Sun Drop Flower | 465 | 201 | 162 | 1435 |
| Sun Drop Flower | 576 | 488 | 440 | 1130 |
| Sun Drop Flower | 390 | 388 | 199 | 1171 |
| Sun Drop Flower | 164 | 115 | 324 | 554 |
| Sun Drop Flower | 790 | 578 | 449 | 530 |
If this table is in excel, we would want to export it as tab-delimited
file in order to further manipulate using UNIX commands and then import
it in R. For exporting: File > Save As > choose format
Text (Tab delimited)(*.txt) and Save. Manipulating with UNIX
commands: open the Lixnux sub-system (Windows) or Termina (Mac/Linux)
and navigate to the directory the file is saved. If you are in Windows,
you will need to run dos2unix to convert the line endings correctly.
To format the above table to just 3 columns i.e,:
1
Organism family length
Now we are ready to manipulate in R!
First, open the R prompt by typing R in the terminal, navigate the
working directory, where the
mythical_creatures.txt file is saved.
1
2
3
R
#then
setwd("path/to/your/directory")
Import data and examine contents, format it if necessary:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
file <- "data/mythical_creatures.txt" # Path to input file or "formatted.txt"
input <- read.csv(file,
sep = "\t",
quote = '',
stringsAsFactors = TRUE,
header = TRUE)
#examine
head(input)
#> Organism gene_family_1 gene_family_2 gene_family_3 gene_family_4
#> 1 Unicorn 263 407 460 519
#> 2 Unicorn 269 644 409 904
#> 3 Unicorn 705 592 489 831
#> 4 Unicorn 501 368 245 1269
#> 5 Unicorn 244 520 403 227
#> 6 Unicorn 387 275 280 185
# Check the header and rename the headers to something shorter
names(input)
#> [1] "Organism" "gene_family_1" "gene_family_2" "gene_family_3"
#> [5] "gene_family_4"
names(input) <- c("Organism","fam1","fam2", "fam3", "fam4")
head(input)
#> Organism fam1 fam2 fam3 fam4
#> 1 Unicorn 263 407 460 519
#> 2 Unicorn 269 644 409 904
#> 3 Unicorn 705 592 489 831
#> 4 Unicorn 501 368 245 1269
#> 5 Unicorn 244 520 403 227
#> 6 Unicorn 387 275 280 185
We will convert the data to tidy data:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
# install.packages("tidyr") # <= in case you need to install tidyr library first
input <- tidyr::pivot_longer(
input,
cols = -Organism,
names_to = "family",
values_to = "length"
)
head(input)
#> # A tibble: 6 x 3
#> Organism family length
#> <fct> <chr> <int>
#> 1 Unicorn fam1 263
#> 2 Unicorn fam2 407
#> 3 Unicorn fam3 460
#> 4 Unicorn fam4 519
#> 5 Unicorn fam1 269
#> 6 Unicorn fam2 644
If your data looks like this below, you’re all set:
1
2
3
4
5
6
7
8
9
> head(input)
Organism family length
1 Unicorn fam1 263
2 Unicorn fam1 269
3 Unicorn fam1 705
4 Unicorn fam1 501
5 Unicorn fam1 244
6 Unicorn fam1 387
>
First, lets load all the required packages. We will need the ggplot2
for plotting, grid package for making grids of plots/faceting.
1
2
library(ggplot2)
library(grid)
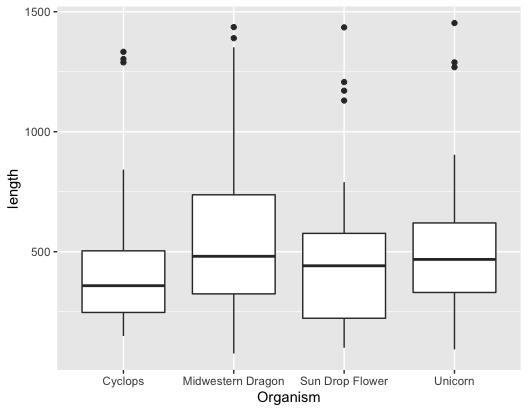
For getting a simple boxplot, you can use the following command:
1
ggplot(input, aes(x = Organism, y = length)) + geom_boxplot()
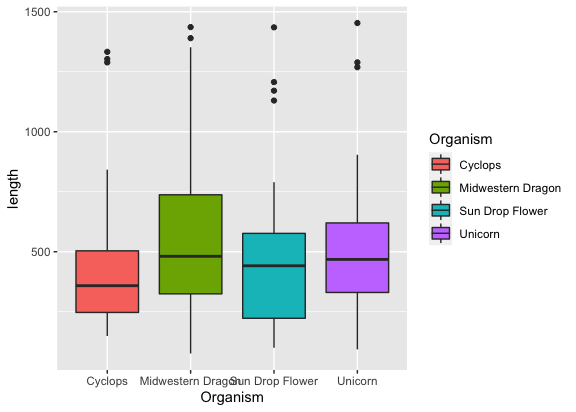
if you want to color the plots,
1
ggplot(input, aes(x = Organism, y = length, fill = Organism)) + geom_boxplot()
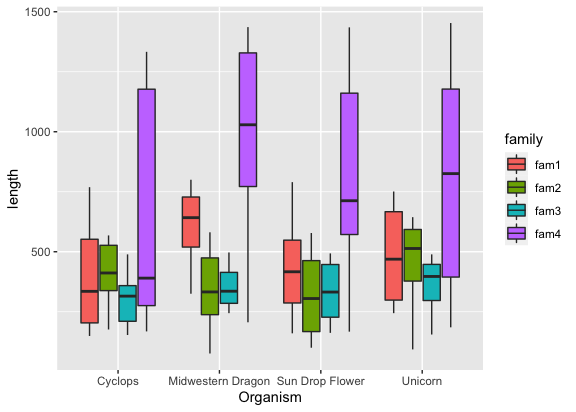
but wait, where are the gene families?
1
ggplot(input, aes(x = Organism, y = length, fill = family)) + geom_boxplot()
That names are all overlapping though?
1
2
3
ggplot(input, aes(x = Organism, y = length, fill = family)) +
geom_boxplot() +
theme(axis.text.x = element_text(angle = 45, hjust=1))
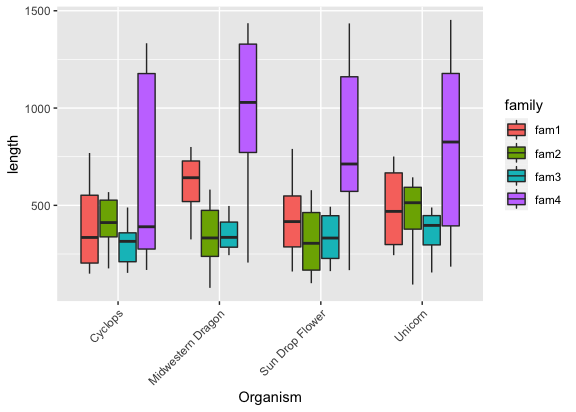
Well, it seems like it is difficult to compare as all gene families are together, can we spread them out in to separate plots?
Yes, but first we need to make different subsets that we want to plot
1
2
3
4
fam1 <- input[which(input$family == "fam1"),]
fam2 <- input[which(input$family == "fam2"),]
fam3 <- input[which(input$family == "fam3"),]
fam4 <- input[which(input$family == "fam4"),]
Let’s also make colors to fill in for each family
1
2
3
4
5
6
7
8
fill1 <- "olivedrab1"
lines1 <- "olivedrab4"
fill2 <- "plum2"
lines2 <- "purple1"
fill3 <- "gold"
lines3 <- "goldenrod3"
fill4 <- "chartreuse"
lines4 <- "chartreuse4"
plots for each family can be generated using dataframes for respective families:
1
2
3
4
ggplot(fam1, aes(x=Organism,y=length)) + geom_boxplot(colour = lines1, fill = fill1)
ggplot(fam2, aes(x=Organism,y=length)) + geom_boxplot(colour = lines2, fill = fill2)
ggplot(fam3, aes(x=Organism,y=length)) + geom_boxplot(colour = lines3, fill = fill3)
ggplot(fam4, aes(x=Organism,y=length)) + geom_boxplot(colour = lines4, fill = fill4)
Instead of generating individual plots, we can create a combined plots,
arranged based on families using facet_grid:
Vertically
1
2
3
4
ggplot(input, aes(x = Organism, y = length, fill = Organism)) +
geom_boxplot() +
theme(axis.text.x = element_text(angle = 45, hjust=1)) +
facet_grid( family ~.) # <= facet allows plots to be split by group
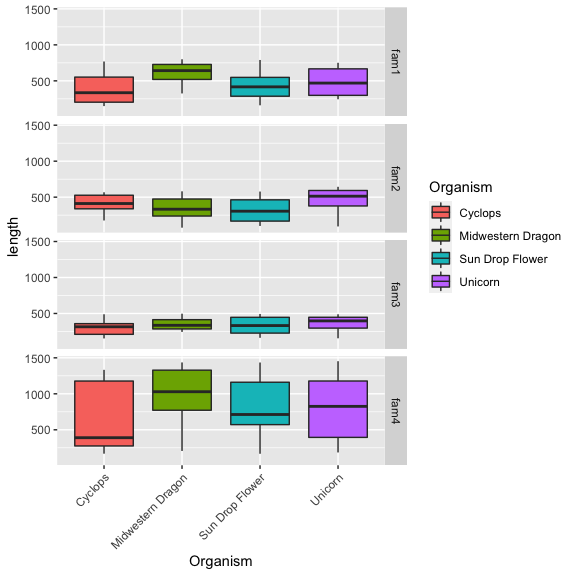
Horizontally
1
2
3
4
ggplot(input, aes(x = Organism, y = length, fill = Organism)) +
geom_boxplot() +
theme(axis.text.x = element_text(angle = 45, hjust=1)) +
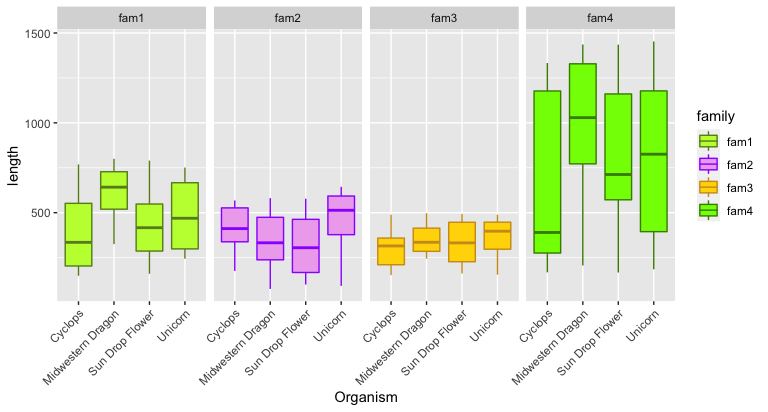
facet_grid( . ~ family) # <= swap the dot and family to arrange horizontally
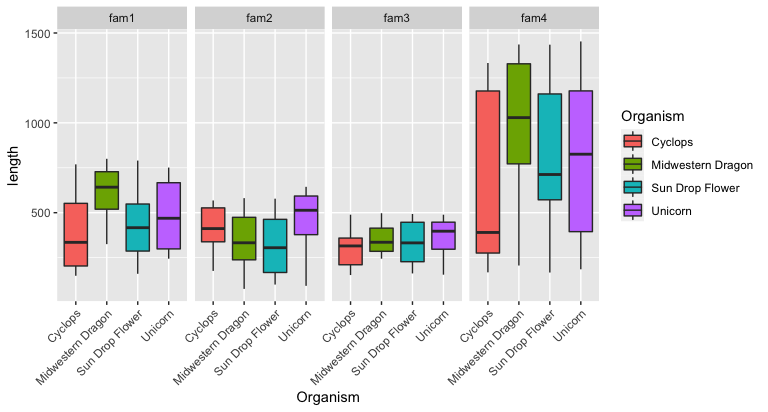
To color these gene families, create a color palette:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
# Fill Color
fam_palette = c(fam1 = "olivedrab1",
fam2 = "plum2",
fam3 = "gold",
fam4 = "chartreuse")
# Line Color
fam_palette2 = c(fam1 = "olivedrab4",
fam2 = "purple1",
fam3 = "goldenrod3",
fam4 = "chartreuse4")
ggplot(input, aes(x = Organism, y = length, fill = family, color = family)) +
geom_boxplot() +
theme(axis.text.x = element_text(angle = 45, hjust=1)) +
facet_grid( . ~ family) +
scale_fill_manual(values = fam_palette) + # <= Add custom color palette here
scale_color_manual(values = fam_palette2)