
Learning Objectives
- Introduction
- Lesson 1: Nextflow params and variables
- Lesson 2: Nextflow Config file
- Lesson 3: Nextflow process
- Lesson 4: Integrating params with process
- Lesson 5: Adding more params
- Lesson 6: How far can we take params?
- Lesson 7: Adding a help param
- Lesson 8: Nextflow process input and output
- Lesson 9: Nextflow channels and process inputs.
- Lesson 10: Nextflow split fasta
- Lesson 11: Advanced Nextflow config
- Lesson 12: nextflow containers
- Lesson 13: makeBlastDB process
- Lesson 14: Resources
Introduction
Presumably, you are here because you want to take your bash scripts and put them into a functioning workflow and have all the execution taken care of for you. You have come to the right place. Nextflow is great but if you are a biologist turned informatician, the groovy language might trip you up a little in many of the great documentation materials that can be found here:
This is a great resource, but it assumes you have had some experience with object oriented programming or even some background in groovy/Java. You will run across methods that aren’t part of nextflow but part of the groovy language like [.trim, .flatten, and the word "it"]. At first this was hard to separate out when we just wanted to know how to do X. While they do have examples in their nextflow patterns section, it isn’t quite sufficient yet to quickly learn to create a nextflow workflow.
Seqera labs just came out (End of August 2020) with a very comprehensive set of tutorials and I strongly encourage you to explore it as it covers everything you will need to create nextflow workflows. However, if you are looking for a shorter, quick start guide type tutorial geared toward biologist turned bioinformatician, this tutorial is for you.
A practical example
The goal of this tutorial is to introduce you to the concepts of nextflow by building a practical example. We will journey through the process of making a simple blast workflow that can take in a fasta file as a query and run blast on it. We will then keep extending this example to showcase different features in nextflow that are useful in building a dynamic workflow.
Prerequisites
This tutorial assumes that you are familiar with bash scripting and how to run blast locally. And perhaps how to use Github.
Nextflow setup
Create a github repo
Let’s start by setting up our folder. I usually do this by first creating a github repo and then cloning it so that I can use version control. I am going to create this in the isugifNF organization that I am apart of and name it tutorial. You should make a repo on your own account.
Then pull it to my local machine. Do not pull this repo as it will download the entire finished tutorial.
1
git clone git@github.com:isugifNF/tutorial.git
Files
Every nextflow workflow requires two main files.
- main.nf
- The main.nf file contains the main nextflow script that calls the processes.
- It doesn’t have to be named main.nf but that is standard practice.
- nextflow.config
- The config file contains default parameters to use in the nextflow pipeline
1
touch main.nf nextflow.config
Your folder should now contain the following
1
2
ls
README.md main.nf nextflow.config
Lesson 1: Nextflow params and variables
Let’s create a simple program that has a parameter to set our query input file that we will use for the BLAST program.
1
2
3
4
5
6
#! /usr/bin/env nextflow
blastdb="myBlastDatabase"
params.query="file.fasta"
println "I will BLAST $params.query against $blastdb"
Let’s dissect it line by line.
The first line is required for all nextflow programs
1
#! /usr/bin/env nextflow
The second line of code sets a variable inside the nextflow script.
1
blastdb="myBlastDatabase"
The third line of code sets a pipeline parameter that can be set at the command line, which I will show you in just a minute. If you want to make a variable a pipeline parameter just prepend the variable with params.
1
params.query="file.fasta"
The last line is a simple print statement that uses both a nextflow variable and a pipeline parameter.
1
println "I will BLAST $params.query against $blastdb"
Go ahead and run it.
1
nextflow run main.nf
Output should look like this
1
2
3
N E X T F L O W ~ version 20.07.1
Launching `main.nf` [confident_williams] - revision: f407a6b0e1
I will BLAST file.fasta against myBlastDatabase
You will also have the default work/ folder that will appear but will be empty as we didn’t do anything but print something to standard out.
Exercises for comprehension
- Pipeline parameters can be set on the command line.
1
nextflow run main.nf --query "newQuery.fasta"
Output:
1
2
3
N E X T F L O W ~ version 20.07.1
Launching `main.nf` [extravagant_pasteur] - revision: f407a6b0e1
I will BLAST newQuery.fasta against myBlastDatabase
- Nextflow script variables cannot be set at the command line
1
nextflow run main.nf --blastdb "NewBlastDB"
Output:
1
2
3
N E X T F L O W ~ version 20.07.1
Launching `main.nf` [loving_borg] - revision: f407a6b0e1
I will BLAST file.fasta against myBlastDatabase
- Try other –param.query inputs on your own.
1
nextflow run main.nf --query "changethistext"
Github saving and ignoring
1
2
3
git add main.nf nextflow.config
git commit -c "started a new nextflow project!"
git push origin master
Add the following to your .gitignore file
1
2
3
.nextflow*
work
out_dir
Then save it to your repo
1
2
3
git add .gitignore
git commit -c "added .gitignore"
git push origin master
Lesson 2: Nextflow Config file
The nextflow config file is nextflow.config. In here, we can set default global parameters for pipeline parameters (params), process, manifest, executor, profiles, docker, singularity, timeline, report and more.
For now, we are going to just add additional pipeline parameters and move the params.query out of the main.nf file and into the nextflow.config file.
Inside the nextflow.config file add the following parameters.
nextflow.config
1
2
3
4
5
params.query = "myquery.fasta"
params.dbDir = "/path/to/my/blastDB/"
params.dbName = "myBlastDB"
params.threads = 16
params.outdir = "out_dir"
Remove these lines from main.nf
1
2
blastdb="myBlastDatabase"
params.query="file.fasta"
and let’s modify the last print statement to include all the parameters.
1
println "I want to BLAST $params.query to $params.dbDir/$params.dbName using $params.threads CPUs and output it to $params.outdir"
Your main.nf file should look like this.
1
2
3
4
/usr/bin/env nextflow
println "I want to BLAST $params.query to $params.dbDir/$params.dbName using $params.threads CPUs and output it to $params.outdir"
output:
1
2
3
4
nextflow run main.nf
N E X T F L O W ~ version 20.07.1
Launching `main.nf` [fervent_swanson] - revision: 418bbdfbef
I want to BLAST myquery.fasta to /path/to/my/blastDB//myBlastDB using 16 CPUs and output it to out_dir
Let’s add a \n to the beginning and end of the print statement so it reports a little more cleanly
1
2
3
4
/usr/bin/env nextflow
println "\nI want to BLAST $params.query to $params.dbDir/$params.dbName using $params.threads CPUs and output it to $params.outdir\n"
output: This looks better:
1
2
3
4
5
6
nextflow run main.nf
N E X T F L O W ~ version 20.07.1
Launching `main.nf` [modest_crick] - revision: 87c6232474
I want to BLAST myquery.fasta to /path/to/my/blastDB//myBlastDB using 16 CPUs and output it to out_dir
Alternate params config
We can also write the pipeline parameters in a different format that is more similar to what we will be using for the rest of the config definitions.
Instead of
1
2
3
4
5
params.query = "myquery.fasta"
params.dbDir = "/path/to/my/blastDB/"
params.dbName = "myBlastDB"
params.threads = 16
params.outdir = "out_dir"
We can write it as follows. Go ahead and change the nextflow.config file to look like this and rerun it to verify to yourself that it works identically.
1
2
3
4
5
6
7
params {
query = "myquery.fasta"
dbDir = "/path/to/my/blastDB/"
dbName = "myBlastDB"
threads = 16
outdir = "out_dir"
}
You will still need to refer to these parameters as params.X.
1
nextflow run main.nf
Lesson 3: Nextflow process
Process definitions are what nextflow uses to define a script to run, the input to the script and the output of a script. Ultimately workflows are comprised of nextflow processes. In this example, we want to run BLAST.
Setup and input files
We will need a fasta file and a database. We are going to use a toy example with only five fasta reads and use that input to generate the blast database and then align it to itself.
Copy and paste the following fasta reads into a file named input.fasta
input.fasta
>Scaffold_1_1..100 CAGGCAAAATGTGGCACAAAAACAACAAATTGTTTAGTAGATACAGGGGCATCCATTTGTTGTATTTCGTCTGCTTTTCTGAGCACAGCTTTTGAAAACC >Scaffold_1_101..200 TTACTCTTGGAAACTCACCCTTTCCACAGGTAAAAGGTGTTGGCGGCGAATTGCATAAAGTGTTAGGTTCAGTTGTGTTAGATTTTGTCATTGAGGATCA >Scaffold_1_201..300 GGAATTTTCTCAAAGATTCTATGTACTGCCTACACTGCCGAAGGCAGTGATACTAGGTGAGAACTTCCTTAATGACAATGATGCAGTCTTAGATTATAGC >Scaffold_1_301..400 TGTCATTCCTTGATACTCAACAACAGCACCTCAGATAGGCAATATATCAATTTCATAGCCAATTCAGTGCATGAGATTAGTGGATTAGCAAAAACACTAG >Scaffold_1_401..500 ATCAGATTTACATCCCCCCTCAGAGTGAAATTCATTTCAAGGTCAGACTATCAGAGACCAAAGAGGATTCCCTCATCCTCATTGAACCCATTGCTTCCCT
Create the BLASTDB
You will need to have BLAST-plus installed on your computer. We will use the makeblastdb command to create a nucleotide database of the input.fasta file and then move the output to a folder named DB
1
2
3
makeblastdb -in input.fasta -dbtype 'nucl' -out blastDB
mkdir DB
mv blastDB.n* DB
Test that BLAST works
This is the command we want to get into a process in a nextflow script.
1
blastn -num_threads 2 -db $PWD/DB/blastDB -query $PWD/input.fasta -outfmt 6 -out input.blastout
nextflow runBlast process
To write this as a nextflow process, we would write it in the following way. The command you want to run should be placed between the """
1
2
3
4
5
6
7
8
process runBlast {
script:
"""
blastn -num_threads 2 -db $PWD/DB/blastDB -query $PWD/input.fasta -outfmt 6 -out input.blastout
"""
}
Place this below the println statement in your main.nf script.
Note: it is critical that the input files use the full path. This is why we have the $PWD (path of working directory Unix variable)
output:
1
2
3
4
5
6
7
8
9
nextflow run main.nf
N E X T F L O W ~ version 20.07.1
Launching `main.nf` [special_sinoussi] - revision: 24972a3b60
I want to BLAST myquery.fasta to /path/to/my/blastDB//myBlastDB using 16 CPUs and output it to out_dir
executor > local (1)
[e0/ac0a8e] process > runBlast [100%] 1 of 1 ✔
The input.blastout will be found in the work folder.
1
2
3
4
5
tree work/
work/
`-- e0
`-- ac0a8e23e6db0eb4b4e81b7d1656f5
`-- input.blastout
The folders inside the work directory are named based on a hash that allows nextflow to -resume from wherever it failed or was stopped.
Lesson 4: Integrating params with process
Now that we have a working nextflow script, lets replace all that we can in the BLAST script with pipeline parameters we can set from the command line.
The BLAST script inside main.nf currently looks like this.
1
2
3
4
script:
"""
blastn -num_threads 2 -db $PWD/DB/blastDB -query $PWD/input.fasta -outfmt 6 -out input.blastout
"""
As you recall and can look up in nextflow.config these are the current pipeline default parameters we have defined.
1
2
3
4
5
6
7
params {
query = "myquery.fasta"
dbDir = "/path/to/my/blastDB/"
dbName = "myBlastDB"
threads = 16
outdir = "out_dir"
}
So we can change the BLAST script as follows
1
2
3
4
script:
"""
blastn -num_threads $params.threads -db $params.dbDir/$params.dbName -query $params.query -outfmt 6 -out input.blastout
"""
We should also adjust the defaults in nextflow.config to real files.
1
2
3
4
5
6
7
params {
query = "$PWD/input.fasta"
dbDir = "$PWD/DB/"
dbName = "blastDB"
threads = 2
outdir = "out_dir"
}
Exercise
- Make the changes described above to the main.nf and the nextflow.config scripts and show that it still works with
nextflow run main.nf - Test out the pipeline parameters
--query--threads
1
nextflow run main.nf --query "$PWD/input.fasta" --threads 6
Lesson 5: Adding more params
We can add more functionality to this script
1
2
3
4
script:
"""
blastn -num_threads $params.threads -db $params.dbDir/$params.dbName -query $params.query -outfmt 6 -out input.blastout
"""
We can add the following:
| parameter | description |
|---|---|
| –outfmt | To change the out format of blast |
| –outFileName | to change the outfile name |
| –options | to add additional options like evalue |
This is what the new script will look like.
1
2
3
4
script:
"""
blastn -num_threads $params.threads -db $params.dbDir/$params.dbName -query $params.query -outfmt $params.outfmt $params.options -out $params.outFileName
"""
Of course if we do that we also need to add it to the nextflow.config file.
1
2
3
4
5
6
7
8
9
10
params {
query = "$PWD/input.fasta"
dbDir = "$PWD/DB/"
dbName = "blastDB"
threads = 2
outdir = "out_dir"
outFileName = "input.blastout"
options = "-evalue 1e-3"
outfmt = "'6'"
}
- Make the changes described above to the main.nf and the nextflow.config scripts and show that it still works with
nextflow run main.nf - Test out the pipeline parameters
--outFileName--optionsand--outfmt
example
1
nextflow run main.nf --outFileName "myBlast" --options "-evalue 1e-10" --outfmt "'6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore qlen slen frames salltitles qcovs'"
Lesson 6: How far can we take params?
- adding an app param.
Turns out we can even add a parameter for the BLAST app we want to run. Right now it only runs blastn but what about blastp,tblastn and blastx? We could use a series of if statements or we can add a params.app that is replaced in the script itself like this.
1
2
3
4
script:
"""
$params.app -num_threads $params.threads -db $params.dbDir/$params.dbName -query $params.query -outfmt $params.outfmt $params.options -out $params.outFileName
"""
Be sure to add the –app in the nextflow.config file.
1
2
3
4
5
6
7
8
9
10
11
params {
query = "$PWD/input.fasta"
dbDir = "$PWD/DB/"
dbName = "blastDB"
threads = 2
outdir = "out_dir"
outFileName = "input.blastout"
options = "-evalue 1e-3"
outfmt = "'6'"
app = "blastn"
}
test it
1
nextflow run main.nf --outFileName "myBlast" --options "-evalue 1e-10" --outfmt "'6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore qlen slen frames salltitles qcovs'" --app tblastx
How do you know if it worked as you expected?
In the work folder in each of the leaf directories you will find a bunch of .command.X files.
1
2
3
4
5
6
7
-rw-r--r-- 1 severin staff 0B Aug 28 09:25 work/78/1c37092ffb6cd83a4973e8f64bc398/.command.begin
-rw-r--r-- 1 severin staff 0B Aug 28 09:25 work/78/1c37092ffb6cd83a4973e8f64bc398/.command.err
-rw-r--r-- 1 severin staff 0B Aug 28 09:25 work/78/1c37092ffb6cd83a4973e8f64bc398/.command.log
-rw-r--r-- 1 severin staff 0B Aug 28 09:25 work/78/1c37092ffb6cd83a4973e8f64bc398/.command.out
-rw-r--r-- 1 severin staff 2.5K Aug 28 09:25 work/78/1c37092ffb6cd83a4973e8f64bc398/.command.run
-rw-r--r-- 1 severin staff 328B Aug 28 09:25 work/78/1c37092ffb6cd83a4973e8f64bc398/.command.sh
-rw-r--r-- 1 severin staff 1B Aug 28 09:25 work/78/1c37092ffb6cd83a4973e8f64bc398/.exitcode
- The
.command.beginis the begin script if you have one (we don’t in this one) - The
.command.erris useful when it crashes. - The
.command.runis the full nextflow pipeline that was run, this is helpful when trouble shooting a nextflow error rather than the script error. - The
.command.shshows what was run. - The
.exitcodewill have the exit code in it.
The .command.sh command will have the command that was actually run. As you can see params.app was successfully replaced with tblastx.
1
2
3
4
cat work/78/1c37092ffb6cd83a4973e8f64bc398/.command.sh
#!/bin/bash -ue
tblastx -num_threads 2 -db /Users/severin/nextflow/workbook/blast/tutorial/DB//blastDB -query /Users/severin/nextflow/workbook/blast/tutorial/input.fasta -outfmt '6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore qlen slen frames salltitles qcovs' -evalue 1e-10 -out myBlast
Lesson 7: Adding a help param
With all these pipeline parameters it is getting hard to remember what we can use. So let’s make a handy help function and --help parameter.
Define a function
To define a function in nextflow we use this type of syntax.
1
2
3
def helpMessage() {
}
We are going to add a help message. The log.info command is not well documented but can be used to print a multiline information using groovy’s logger fuctionality. In simple terms put what you want to print out for the help message between the log.info """ and """
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
def helpMessage() {
log.info """
Usage:
The typical command for running the pipeline is as follows:
nextflow run main.nf --query QUERY.fasta --dbDir "blastDatabaseDirectory" --dbName "blastPrefixName"
Mandatory arguments:
--query Query fasta file of sequences you wish to BLAST
--dbDir BLAST database directory (full path required)
--dbName Prefix name of the BLAST database
Optional arguments:
--outdir Output directory to place final BLAST output
--outfmt Output format ['6']
--options Additional options for BLAST command [-evalue 1e-3]
--outFileName Prefix name for BLAST output [input.blastout]
--threads Number of CPUs to use during blast job [16]
--chunkSize Number of fasta records to use when splitting the query fasta file
--app BLAST program to use [blastn;blastp,tblastn,blastx]
--help This usage statement.
"""
}
Run a the function
Now that the definition has been defined above, we need code that executes it if the params.help parameter is set.
Comments are specified in groovy with //
1
2
3
4
5
// Show help message
if (params.help) {
helpMessage()
exit 0
}
the exit 0 tells nextflow to quit running and exit after printing the help message.
-
Paste the above helpMessage() function and if statement in the
main.nffile aboveprocess runBlast.main.nf
#! /usr/bin/env nextflow println "\nI want to BLAST $params.query to $params.dbDir/$params.dbName using $params.threads CPUs and output it to $params.outdir\n" def helpMessage() { log.info """ Usage: The typical command for running the pipeline is as follows: nextflow run main.nf --query QUERY.fasta --dbDir "blastDatabaseDirectory" --dbName "blastPrefixName" Mandatory arguments: --query Query fasta file of sequences you wish to BLAST --dbDir BLAST database directory (full path required) --dbName Prefix name of the BLAST database Optional arguments: --outdir Output directory to place final BLAST output --outfmt Output format ['6'] --options Additional options for BLAST command [-evalue 1e-3] --outFileName Prefix name for BLAST output [input.blastout] --threads Number of CPUs to use during blast job [16] --chunkSize Number of fasta records to use when splitting the query fasta file --app BLAST program to use [blastn;blastp,tblastn,blastx] --help This usage statement. """ } // Show help message if (params.help) { helpMessage() exit 0 } process runBlast { script: """ $params.app -num_threads $params.threads -db $params.dbDir/$params.dbName -query $params.query -outfmt $params.outfmt $params.options -out $params.outFileName """ } -
then add the pipeline parameter
help = falseto your params code block innextflow.config.nextflow.config
params { query = "$PWD/input.fasta" dbDir = "$PWD/DB/" dbName = "blastDB" threads = 2 outdir = "out_dir" outFileName = "input.blastout" options = "-evalue 1e-3" outfmt = "'6'" app = "blastn" help = false }
Exercise
- Try it out and make sure it works.
1
nextflow run main.nf --help
Lesson 8: Nextflow process input and output
So far we have written a nextflow script and replaced as much of it as we could with pipeline parameters and learned how to write a function in nextflow to display a usage message. Now we are going to extend the process to include input and output sections.
Current runBlast process
1
2
3
4
5
6
process runBlast {
script:
"""
$params.app -num_threads $params.threads -db $params.dbDir/$params.dbName -query $params.query -outfmt $params.outfmt $params.options -out $params.outFileName
"""
}
Adding nextflow output and publishDir sections
Here we have added a nextflow function called publishDir which tells nextflow where to place a symlink of the output files. Those outputfiles are defined by the output: section of the process.
Note: the params.outdir in the publishDir command has a $ in front of it because it is inside a quotation whereas the params.outFileName does not have a $ because it is not inside a script or a set of quotes. This is a common source of errors when first scripting in nextflow. Params do not work inside single quote statements as far as I can tell with or without a dollar sign.
You can have more than one line in the output: section. It makes sense to put the publishDir in the output section.
1
2
3
4
5
6
7
8
9
10
11
process runBlast {
output:
publishDir "${params.outdir}/blastout"
path(params.outFileName)
script:
"""
$params.app -num_threads $params.threads -db $params.dbDir/$params.dbName -query $params.query -outfmt $params.outfmt $params.options -out $params.outFileName
"""
}
output
You should now see an out_dir directory with a blastout subdirectory and the input.blastout in side that folder.
Note: it is important that the the files specified in the output match any pattern you specify in publishDir. see More complicated example below.
More complicated example
This is an output section from a busco workflow I have been working on. As you can see, I define the output file I want to copy over into the `publishDir` I have to specify the folder `${label}` in both the file output and the publishDir patthern location for it to work properly.
```
output:
file("${label}/short_summary.specific.*.txt")
publishDir "${params.outdir}/BUSCOResults/${label}/", mode: 'copy', pattern: "${label}/short_summary.specific.*.txt"
```
Lesson 9: Nextflow channels and process inputs.
Turns out nextflow inputs are intrinsically linked to nextflow channels. So I can’t talk about inputs without speaking about channels. Process inputs are taken from a nextflow channel.
Channels can act as either a queue or value.
- queue channels are consumable. A channel is loaded with a file and a process will use the file and empty the channel of that file.
- value channels are not consumed. A channel is loaded with a value and a process or several processes can use that channel value over and over again.
With Channels, we will start to see more nextflow operators for channels. In this case, we are going to add the following code above the runBlast process which states create a channel from a file path and set the channel name into queryFile_ch.
Note: Operators may take the method syntax () or the closure syntax { }. We see both in this example. This is a common source of errors when writing nextflow scripts.
1
2
3
Channel
.fromPath(params.query)
.into { queryFile_ch }
We can now add an input: section to the process like this
1
2
input:
path(queryFile) from queryFile_ch
And we can swap out $params.query with $queryFile. Some of the readers may be wondering why we would do this if it does the exactly the same thing? The answer is that with processes that can accept a channel as input we have greater control to parallelize the execution of the process on the data as we will see in the next lesson.
main.nf
process runBlast {
input:
path queryFile from queryFile_ch
output:
publishDir "${params.outdir}/blastout"
path(params.outFileName)
script:
"""
$params.app -num_threads $params.threads -db $params.dbDir/$params.dbName -query $queryFile -outfmt $params.outfmt $params.options -out $params.outFileName
"""
}
Run it and verify it works
1
nextflow run main.nf
Lesson 10: Nextflow split fasta
One of the more powerful channel operators is called .splitFasta. This operator can be used to split a fasta file into chunks by a specified chunk size and then we can pass it to the queue and the process will run these chunks in parallel.
To do this, we modify the queryFile_ch Channel. This code says to create a channel from path params.query and splitFasta by chunks of size 1 fasta record and make a file for these chunks in the work process folder and put this into a channel named queryFile_ch. I chose a chunk size of 1 because our example fasta only has 5 fasta records
1
2
3
4
Channel
.fromPath(params.query)
.splitFasta(by: 1, file:true)
.into { queryFile_ch }
When you run this you will now see a different output
1
2
3
4
5
6
7
nextflow run main.nf
N E X T F L O W ~ version 20.07.1
Launching `main.nf` [suspicious_kare] - revision: df99b16d40
WARN: The `into` operator should be used to connect two or more target channels -- consider to replace it with `.set { queryFile_ch }`
executor > local (5)
[e1/5baaab] process > runBlast (4) [100%] 5 of 5 ✔
The important part of this is that there are now 5 local jobs and 5 runBlast processes that have been performed. You will also see more folders in your work folder.
PublishDir broke
Something that you may have noticed is that the input.blastout now only contains a single blast line. What has happened is that every time the runBlast process runs it replaces the BLAST output of the fasta chunk in publishDir. To fix this we need to send the output of all these chunks to a new channel and then use a different parameter to collect them before publishing.
So change the runBlast process output line to look like this.
1
2
output:
path(params.outFileName) into blast_output_ch
Notice that you will see the into method again and then the name of the channel you are creating.
After the runBlast process add the following lines.
1
2
blast_output_ch
.collectFile(name: 'blast_output_combined.txt', storeDir: params.outdir)
This takes the blast_output_ch that has the blast output from the fasta chunks and collects them (cat) into a new file named blast_output_combined.txt in the folder named params.outdir.
Output
The out_dir now contains the blast_output_combined.txt file with all the blast output.
1
2
out_dir/
blast_output_combined.txt blastout
If you messed up or need help, this is what your main.nf script should now look like
main.nf
#! /usr/bin/env nextflow
println "\nI want to BLAST $params.query to $params.dbDir/$params.dbName using $params.threads CPUs and output it to $params.outdir\n"
def helpMessage() {
log.info """
Usage:
The typical command for running the pipeline is as follows:
nextflow run main.nf --query QUERY.fasta --dbDir "blastDatabaseDirectory" --dbName "blastPrefixName"
Mandatory arguments:
--query Query fasta file of sequences you wish to BLAST
--dbDir BLAST database directory (full path required)
--dbName Prefix name of the BLAST database
Optional arguments:
--outdir Output directory to place final BLAST output
--outfmt Output format ['6']
--options Additional options for BLAST command [-evalue 1e-3]
--outFileName Prefix name for BLAST output [input.blastout]
--threads Number of CPUs to use during blast job [16]
--chunkSize Number of fasta records to use when splitting the query fasta file
--app BLAST program to use [blastn;blastp,tblastn,blastx]
--help This usage statement.
"""
}
// Show help message
if (params.help) {
helpMessage()
exit 0
}
Channel
.fromPath(params.query)
.splitFasta(by: 1, file:true)
.into { queryFile_ch }
process runBlast {
input:
path queryFile from queryFile_ch
output:
publishDir "${params.outdir}/blastout"
path(params.outFileName) into blast_output_ch
script:
"""
$params.app -num_threads $params.threads -db $params.dbDir/$params.dbName -query $queryFile -outfmt $params.outfmt $params.options -out $params.outFileName
"""
}
blast_output_ch
.collectFile(name: 'blast_output_combined.txt', storeDir: params.outdir)
Summary
Up to this point you know have a working runBlast workflow that will take in a fasta file split it into chunks of size 1 fasta record and run those blasts in parallel and then collect all the outputs and write it to a single file in the out_dir.
Exercise
- Add a chunkSize parameter that you can set so that you can specify the chunksize. Be sure to add it to the appropriate places in nextflow.config and main.nf
Hints
1. params has an s at the end of it 2. It is not contained inside `"` so don't use the `$`
Lesson 11: Advanced Nextflow config
There is quite a bit to learn about the nextflow config file that adds significant functionality and modularity to nextflow. Here are some highlights.
timeline
If the following code is added to your nextflow.config file, nextflow will output a detailed timeline report.
-
nextflow.config
1 2 3 4
timeline { enabled = true file = "$params.outdir/timeline.html" }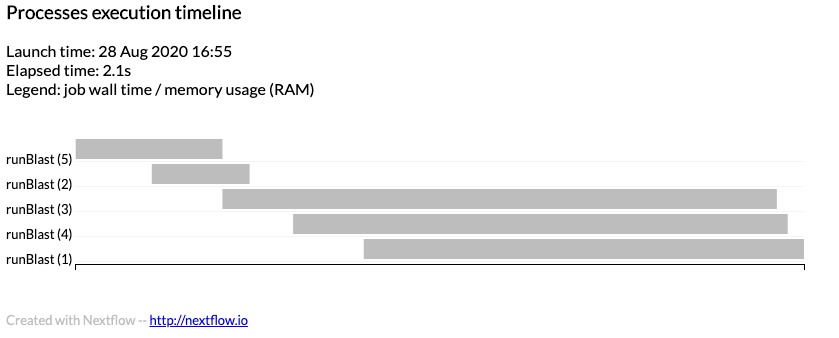
report
If the following code is added to your nextflow.config file, nextflow will output a detailed execution report.
-
nextflow.config
1 2 3 4
report { enabled = true file = "$params.outdir/report.html" }The report has technical detail about cpu and memory usage for each process but this toy example is too small to show anything interesting.
executors
The executor code block in the nextflow.config file can be used to set default executor parameters. In this case, we are setting the queueSize to be no more than 100 jobs and a submission rate of no more than 10 per second.
These parameters are only active if we choose a non-local executor like slurm or torque.
-
nextflow.config
1 2 3 4
executor { queueSize = 100 submitRateLimit = '10 sec' }
profiles
We can also add profiles into the nextflow.config file that will load different settings depending on the profile. This is similar to an include statement in other languages.
-
nextflow.config
1 2 3
profiles { slurm { includeConfig './configs/slurm.config' } } -
./configs/slurm.config
Make a file named slurm.config in a folder named configs and add the following text.
1 2 3 4
process { executor = 'slurm' clusterOptions = '-N 1 -n 16 -t 24:00:00' }
With a profile added you can now execute the specific parameters in a profile by using the -profile nextflow parameters (again this is a nextflow parameter and not a pipeline parameter so it only has a single - just like -resume)
1
nextflow run main.nf -profile slurm
Note: this will fail unless you are on a machine that has sbatch.
Manifest
-
nextflow.config
1 2 3 4 5 6 7 8
manifest { name = 'isugifNF/tutorial' author = 'Andrew Severin' homePage = 'www.bioinformaticsworkbook.org' description = 'nextflow bash' mainScript = 'main.nf' version = '1.0.0' }
You don’t actually need this to run from a github repo but you can use it to specify the mainScript. Default is main.nf.
To execute from a repository you can refer to it as follows.
1
nextflow run isugifNF/tutorial --help
Note: isugifNF/tutorial is my organization/githubRepo, that I have been pushing this tutorial to. It will be different for you.
labels
It is possible to add a label to a process between the process name and the input:.
1
label 'blast'
The label can define in the nextflow.config in your process directive or in our case in the Configs/slurm.config. It can be used like a reusable variable to include additional parameters.
1
2
3
4
5
6
process {
executor = 'slurm'
clusterOptions = '-N 1 -n 16 -t 02:00:00'
withLabel: blast { module = 'blast-plus' }
}
In the above example we have provided code that will load the appropriate module. It would be possible to create several profiles to load the correct modules depending on the super computer you were using, for example. Depending on your super computer you may need to change the name of the module you want to load.
modules
If you want to just load the module directly in the process you can just add the module command like directly where you put the label which is anywhere before the input:
1
2
3
4
5
6
7
8
9
10
process runBlast {
module = 'blast-plus'
publishDir "${params.outdir}/blastout"
input:
path queryFile from queryFile_ch
.
.
. // these three dots mean I didn't paste the whole process.
Lesson 12: nextflow containers
Singularity and Docker
There are default profiles for singularity -profile singularity and docker -profile docker that can be turned on in the nextflow.config file by adding these lines to your profile directive.
1
2
3
4
5
6
7
8
9
10
11
profiles {
slurm { includeConfig './configs/slurm.config' }
docker { docker.enabled = true }
singularity {
singularity.enabled = true
singularity.autoMounts = true
}
}
To have a container run for a process when the above profile is specified a line can be added to the process with the container name as it appears in docker hub
Docker is the default so you can use this short version.
1
container = 'ncbi/blast'
Or you can specify a container using its full path from other locations such as Red Hat Quay.io
1
container = `quay.io/biocontainers/blast/2.2.31--pl526he19e7b1_5`
You can place this line anywhere before the input: in the process.
process runBlast {
container = 'ncbi/blast'
publishDir "${params.outdir}/blastout"
input:
path queryFile from queryFile_ch
.
.
. // these three dots mean I didn't paste the whole process.
example
The first time you run this may take some time to download the container. After that it should run more quickly.
1
nextflow run main.nf -profile docker
The above line fails because it can’t find the database. Let’s try giving the location explicitly inside a profile. let’s create a test profile. Add the following to a test.config file in the configs folder
params {
query = "${projectDir}/input.fasta"
outdir = './out_dir'
dbDir = "${projectDir}/DB/"
chunkSize = 1 //this is the number of fasta
}
Note: nextflow has a bunch of implicit variables one of which is the projectDir I use above which is where the main workflow script is located. It used to be called baseDir.
Now add this line to your profiles directive inside nextflow.config.
1
2
test { includeConfig './configs/test.config' }
Actually this fails too because even though we used a profile, it is still pointing to the local file system and not the container filesystem. We need a way to pass the database file location directly into the runBlast process without the need of the local path. We need to put everything into channels!
// This channel will grab the folder path and set it into a channel named dbDir_ch
Channel.fromPath(params.dbDir)
.set { dbDir_ch }
// this channel will grab the text from params.dbName. Notice it is just from and not fromPath. nextflow will complain if you try to grab a path from a bit of text.
Channel.from(params.dbName)
.set { dbName_ch }
process runBlast {
container = 'ncbi/blast'
publishDir "${params.outdir}/blastout"
// The input now requires these two additinal inputs from their respective channels. Again dbName is a val not a file/folder path.
input:
path queryFile from queryFile_ch
path dbDir from dbDir_ch
val dbName from dbName_ch
output:
path(params.outFileName) into blast_output_ch
// the script section requires that we change the $params.dbDir and $params.dbName to correspond to the input from channel which we have aptly named dbDir and dbName. Since they are inside triple quotes we need to include the dollar sign
script:
"""
$params.app -num_threads $params.threads -db $dbDir/$dbName -query $queryFile -outfmt $params.outfmt $params.options -out $params.outFileName
"""
}
This runs but there is a problem. It only runs the first chunk.
Try it.
nextflow run main.nf -profile docker,test
N E X T F L O W ~ version 20.07.1
Launching `main.nf` [agitated_becquerel] - revision: 8fd35ffcca
I want to BLAST /Users/severin/nextflow/workbook/blast/tutorial/input.fasta to /Users/severin/nextflow/workbook/blast/tutorial/DB//blastDB using 2 CPUs and output it to ./out_dir
WARN: The `into` operator should be used to connect two or more target channels -- consider to replace it with `.set { queryFile_ch }`
executor > local (1)
[ec/8bddfe] process > runBlast (1) [100%] 1 of 1 ✔
To fix this we need to make the channels into value channels not queue channels. We discussed this many lessons ago. Change these two lines to include the .val at the end.
path dbDir from dbDir_ch.val
val dbName from dbName_ch.val
Now try it again
nextflow run main.nf -profile docker,test
N E X T F L O W ~ version 20.07.1
Launching `main.nf` [focused_hamilton] - revision: 26c03ab400
I want to BLAST /Users/severin/nextflow/workbook/blast/tutorial/input.fasta to /Users/severin/nextflow/workbook/blast/tutorial/DB//blastDB using 2 CPUs and output it to ./out_dir
WARN: The `into` operator should be used to connect two or more target channels -- consider to replace it with `.set { queryFile_ch }`
executor > local (5)
[ab/70c53b] process > runBlast (1) [100%] 5 of 5 ✔
Container Notes
There is a containerOptions variable that can also be passed to pass container options to the container. For example, I needed to bind a directory inside the container so that Augustus would work in the BUSCO container. This is not directly related to the blast workflow we are building but wanted to make a note here for your reference about containerOptions.
1
2
3
4
5
6
7
process runBUSCO {
container = "$busco_container"
containerOptions = "--bind $launchDir/$params.outdir/config:/augustus/config"
.
.
.
Fix Warnings
The warning is saying we shouldn’t have used .into at the beginning to set the first channel.
1
WARN: The `into` operator should be used to connect two or more target channels -- consider to replace it with `.set { queryFile_ch }`
Go ahead and change it to .set { queryFile_ch }. We use .into { fastq_reads_qc; fastq_reads_trim } when we want to make multiple channels for the same input. This gets around that consuming thing when you have multiple programs that need to consume the same input.
Lesson 13: makeBlastDB process
There is one more feature that would make this simple nextflow workflow awesome. That feature would be to automatically generate the BLAST databases for us if we provide it with a --genome parameter.
To do this, we will need to:
- Create a
params.genomeparameter - Create if/else statement where if
--genomeis supplied on the command line it will run a process. - Create a process for
makeblastdb - Connect the channels correctly to pass it on to the
runBlastprocess - Add to help usage statement
1) Create a params.genome parameter
- to
nextflow.config
add the following to the params directive:
1
genome = false
2) create if/else statement
Add an if statement after the queryFile_ch statement
1
2
3
4
5
6
7
8
9
10
Channel
.fromPath(params.query)
.splitFasta(by: params.chunkSize, file:true)
.set { queryFile_ch }
if (params.genome) {
println "It worked"
exit 0
}
Test it
1
2
3
4
5
6
7
nextflow run main.nf --genome test.fasta
N E X T F L O W ~ version 20.07.1
Launching `main.nf` [modest_leakey] - revision: 65c14ec981
I want to BLAST /Users/severin/nextflow/workbook/blast/tutorial/input.fasta to /Users/severin/nextflow/workbook/blast/tutorial/DB//blastDB using 2 CPUs and output it to out_dir
It worked
3) Create a process for makeblastdb
Great, now let’s add the process. We are going to build this up over several section below, wait until the end before you try running it.
The script: part is straight forward as we know how to use makeblastdb.
I went ahead and gave the out directory and name the same as what we need for input in the runBlast process. And since, the params.genome parameter is the genome fasta file, I added that for the -in
1
2
3
4
5
6
7
8
9
10
11
if (params.genome) {
process runMakeBlastDB {
script:
"""
makeblastdb -in ${params.genome} -dbtype 'nucl' -out $dbDir/$dbName
"""
}
println "It worked"
exit 0
}
The problem is that we get the dbDir and dbName from Channels and the pipeline parameters. We need another means of capturing this information. We can use a Channel and grab this information from params.genome. In this case, we are using the other method for creating a Channel were we can define the channel name and then an equal sign like a variable. We use .fromPath and the params.genome pipeline parameter. And new to this lesson is the .map nextflow function which will take the input (file) and create a tuple (multivariable output) of file.simpleName(file prefix), file.parent(directory path) and file(full file path).
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
if (params.genome) {
genomefile = Channel
.fromPath(params.genome)
.map { file -> tuple(file.simpleName, file.parent, file) }
process runMakeBlastDB {
script:
"""
makeblastdb -in ${params.genome} -dbtype 'nucl' -out $dbDir/$dbName
"""
}
println "It worked"
exit 0
}
With that mapping we can now add the input and output.
- Input
- from the genomefile_ch we set the (val, path, file) for the tuple (dbName,dbdir,FILE)
- Output
- The output we are just passing the dbName and dbDir into new channels that are read into the
runBlastprocess
- The output we are just passing the dbName and dbDir into new channels that are read into the
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
if (params.genome) {
genomefile_ch = Channel
.fromPath(params.genome)
.map { file -> tuple(file.simpleName, file.parent, file) }
process runMakeBlastDB {
input:
set val(dbName), path(dbDir), file(FILE) from genomefile_ch
output:
val dbName into dbName_ch
path dbDir into dbDir_ch
script:
"""
makeblastdb -in ${params.genome} -dbtype 'nucl' -out $dbDir/$dbName
"""
}
}
I went ahead and deleted these two lines as they were only useful for testing above.
1
2
println "It worked"
exit 0
4) Connect the channels
The last section, creates the BLAST databases but it doesn’t pass it on to runBlast but is over written by the following lines
1
2
3
4
5
Channel.fromPath(params.dbDir)
.set { dbDir_ch }
Channel.from(params.dbName)
.set { dbName_ch }`
so what we need to do is enclose this into an else clause
1
2
3
4
5
6
7
} else {
Channel.fromPath(params.dbDir)
.set { dbDir_ch }
Channel.from(params.dbName)
.set { dbName_ch }`
}
Now we can test if it works by providing it an input.
1
nextflow run main.nf --genome input.fasta --query input.fasta
Test it with your own data. When you do, don’t forget to set the --chunkSize and --threads
5) Add --genome to help usage statement
- to
main.nf
The first thing we see in main.nf is the help usage function. Let’s go ahead and add the following line to it so it is complete.
1
--genome If specified with a genome fasta file, a BLAST database will be generated for the genome
You can see it for yourself with this command
1
nextflow run main.nf --help
Lesson 14: Resources
Congratulations, if you made it this far, you have successfully built a nextflow workflow for running BLAST. You are now at a point where you can easily explore other workflow examples for inspiration on how to create even more complex workflows.
For inspiration, I highly recommend the following sites.
Also Consider adding syntax highlighting to your vim.
1
2
3
4
vim ~/nextflow-vim;
mkdir ~/.vim/ftdetect; mkdir ~/.vim/ftplugin;
cp ~/nextflow-vim/ftdetect/nextflow.vim ~/.vim/ftdetect;
cp ~/nextflow-vim/syntax/nextflow.vim ~/.vim/ftplugin;
Resource list
- Nextflow getting started
- nextflow patterns section
- nextflow operators
- queue or value channels
- nextflow.config
- Containers