
SNP calling for GBS data using Stacks pipeline
Introduction
Genotyping-by-sequencing (GBS) is a technique that allows to rapidly detect nucleotide variation across the whole genome. This allows to genotype large number of individuals, simultaneously, at a very low cost as this is a reduced representation of the genome. It can work with or without a reference genome, and easily generates large number of useful information for these individuals at population level.
Dataset
In this tutorial, we will use NAM population GBS data generated by Rodgers-Melnick et al.. This GBS data is collected from 5000 RIL individuals belonging to NAM population (25 diverse maize lines crossed to B73). Originally, SNPs were called using B73 V3 genome and the results were used for various purposes. Since there is more latest version of the genome, we can replicate the same analyses, using a different tool (Stacks) and an updated genome. In the next tutorial we will create a tutorial for calling SNPs using Tassel.
The dataset for this GBS is available on NCBI SRP009896 and there are 91 SRRs associated with it. The metadata for these SRRs are available on CyVerse Data Commons, at this location:
1
/iplant/home/shared/panzea/genotypes/GBS/v27/
Specifically, you will need AllZeaGBSv2.7_publicSamples_metadata20140411.xlsx file present in that directory. This file is essential not only to demultiplex the SRR data you download, but also, if you are interested in doing downstream analyses it will tell the sample pedigree information.
Organization
The data is organized as follows:
1
2
3
4
5
6
7
8
9
stacks/
├── 1-data
├── 2-genome
└── 3-stacks
├── a-barcodes
├── b-demux
├── c-alignments
├── d-gstacks
└── e-population
Programs required
- Stacks
- NCBI-edirect
- enaBrowserTools
- BWA
- samtools
- Picard
- VCFTools
Overview
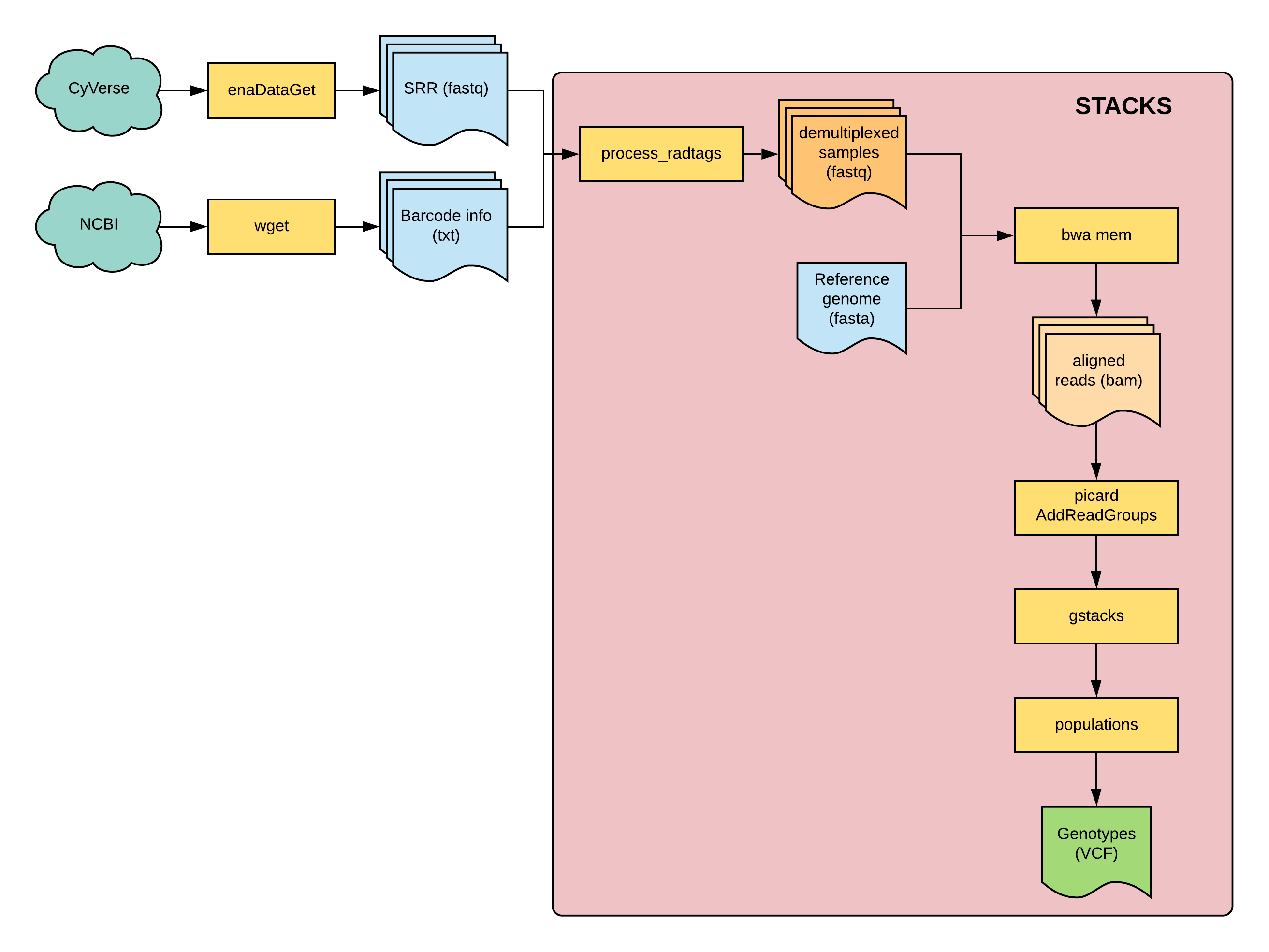 Fig 1: overview of this tutorial
Fig 1: overview of this tutorial
Organize datasets
Fastq
First, download the 91 SRR datasets from the SRA database. For this you can use the NCBI-edirect package to obtain the list of SRRs and then enaBrowserTools to download them.
1
2
3
4
5
6
7
8
9
cd stacks/1-data
esearch -db sra -query SRP009896 |\
efetch --format runinfo |\
cut -d , -f 1 |\
awk 'NF>0' |\
grep -v Run > srr.ids
while read line; do
enaDataGet -f fastq -a $line;
done<srr.ids
This will generate 91 folders with single fastq (compressed) file each.
Barcodes
From the spreadsheet downloaded, you will need to extract the required information (flowcell, lane, barcode, and samplename) and generate a simple text file. You can use the SRR id for the flowcell to make the analyses simpler. If you have the same samplename Since there will be samples with same samplename in multiple SRR files, we need to process them separately. The combined, extracted, and cleaned barcode file is provided here in its final form for reference. If you have your own data, it is essential that you create the barcode file in similar format. This file is placed in 3-stacks directory and it will be split into multiple files based on SRR ids.
1
2
3
4
cd 3-stacks
while read line; do
grep -Fw '$line' barcodes-gbs-data.tsv > a-barcodes/${line}-barcodes.txt;
done<../1-data/srr.ids
Genome
B73 version 4 from Gramene, download and BWA index:
1
2
3
4
5
cd 2-genome
wget ftp://ftp.gramene.org/pub/gramene/CURRENT_RELEASE/fasta/zea_mays/dna/Zea_mays.B73_RefGen_v4.dna.toplevel.fa.gz
gunzip Zea_mays.B73_RefGen_v4.dna.toplevel.fa.gz
module load bwa
bwa index Zea_mays.B73_RefGen_v4.dna.toplevel.fa
Demulitplex
Now, we are ready to start the Stacks pipeline. We will first start by demultiplexing.
1
2
3
4
5
6
module load gcc/7.3.0-xegsmw4
module load stacks
for srr in ../1-data/SRR*; do
ID=$(basename $srr);
echo process_radtags -p ../1-data/${ID}/ -o ./b-demux/${ID}/ -b ./a-barcodes/${ID}-barcodes.txt -e apeKI -r -c -q;
done > process.cmds
Make Slurm scripts and submit makeSLURMp.py 350 process.cmds
1
2
3
4
for sub in process*.sub; do
sed -i 's/parallel -j 1/parallel -j 20/g' $sub;
sbatch $sub
done
Align reads to genome
We will use BWA program for alignments
create a run script runBWA.sh
1
2
3
4
5
6
#!/bin/bash
module load bwa
input=$1
output=$(basename $input | sed 's/.fq.gz/.sam/g')
index=/path/to/stacks/2-genome/Zea_mays.B73_RefGen_v4.dna.toplevel.fa
bwa mem -t 4 ${index} ${input} > c-alignments/${output}
Generate commands and submit:
1
2
3
4
5
6
7
8
for fq in $(find /path/to/stacks/3-stacks/b-demuxed -name "*.gz"); do
echo ./run-bwa.sh $fq;
done > bwa.cmds
makeSLURMp.py 350 bwa.cmds
for sub in bwa*.sub; do
sed -i 's/parallel -j 1/parallel -j 9/g' $sub;
sbatch $sub
done
Alignments will be written to c-alignments directory.
Calling SNPs
For the GBS SNP calling pipeline, you need to have coordinate sorted bam file with readgroups added to the BAM header. We will do this using samtools and picard
script runSort-and-addReadgroups.sh
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
#!/bin/bash
SAM="$1"
samtools view --threads 36 -b -o ${SAM%.*}.bam ${SAM}
samtools sort -o ${SAM%.*}_sorted.bam -T ${SAM%.*}_temp ${SAM%.*}.bam
module load picard
REF="/path/to/stacks/2-genome/Zea_mays.B73_RefGen_v4.dna.toplevel.fa"
ulimit -c unlimited
# example file looks like this:
# Z002E0081-SRR391089_sorted.bam
bam="${SAM%.*}_sorted.bam"
RGID=$(basename $bam | rev | cut -f 1 -d "-" | rev | sed 's/_sorted.bam//g')
RGSM=$(basename $bam | rev | cut -f 2- -d "-" | rev )
RGLB="${RGSM}-L001"
RGPU=001
echo -e "$RGID\t$RGSM\t$RGLB\t$RGPU"
java -Djava.io.tmpdir=$TMPDIR -Xmx50G -jar $PICARD_HOME/picard.jar AddOrReplaceReadGroups \
I=${bam} \
O=${bam%.*}_new.bam \
RGID=$RGSM \
RGLB=$RGLB \
RGPL=ILLUMINA \
RGPU=$RGPU \
RGSM=$RGSM
module load samtools
samtools index ${bam%.*}_new.bam
generate commands and submit:
1
2
3
4
5
6
7
8
for sam in *.sam; do
echo "./runSort-and-addReadgroups.sh $sam";
done > sort.cmds
makeSLURMp.py 350 sort.cmds
for sub in sort*.sub; do
sed -i 's/parallel -j 1/parallel -j 9/g' $sub;
sbatch $sub
done
Next, we run gstacks. This has to be run on all bam files together. So to the command generated will be really long:
1
2
3
for bam in *_new.bam; do
echo " -B $bam \";
done > middle
head file head
1
2
3
4
5
#!/bin/bash
ml purge
ml gcc/7.3.0-xegsmw4
ml stacks
gstacks \
tail file: tail
1
2
-O /path/to/stacks/3-stacks/d-gstacks \
-t 36
combine:
1
2
3
4
rm gstacks.cmds
cat head middle tail >> gstacks.cmds
makeSLURMs.py 100000 gstacks.cmds
sbatch gstacks_0.sub
Finally, run the populations script:
the script: run-populations.sh
1
2
3
4
5
6
7
8
9
10
11
12
13
#!/bin/bash
ml purge
ml gcc/7.3.0-xegsmw4
ml stacks
input="/path/to/stacks/3-stacks/d-gstacks"
output="/path/to/stacks/3-stacks/e-population"
populations \
-P ${input} \
-O ${output} \
-t 36 \
--batch-size 10 \
--vcf \
--phylip
This will generate the final-results in the e-population directory:
1
2
3
4
5
6
7
8
9
10
11
populations.fixed.phylip
populations.fixed.phylip.log
populations.haplotypes.tsv
populations.hapstats.tsv
populations.haps.vcf
populations.log
populations.log.distribs
populations.markers.tsv
populations.snps.vcf
populations.sumstats.tsv
populations.sumstats_summary.tsv