
GATK Best Practices Workflow for DNA-Seq
Dataset
For this tutorial we will use the dataset from BioProject PRJEB18647. This dataset has Illumina short reads for four different populations of Arabidopsis halleri subsp. halleri (Aha18, AhaN1, AhaN3, AhaN4) and was originally used for estimating genomic diversity and population differentiation for these 4 populations (Fischer et. al.,).
Table 1: Dataset used for GATK SNP calling.
| Name | ReadLength | InsertSize | MBases | SRR-id | Genotype |
|---|---|---|---|---|---|
| Aha18 | 202 | 250 | 21,368 | ERR1760144 | A. halleri pop. Aha18 |
| AhaN1 | 200 | 150 | 30,136 | ERR1760145 | A. halleri pop. AhaN1 |
| AhaN3 | 200 | 150 | 29,631 | ERR1760146 | A. halleri pop. AhaN3 |
| AhaN4 | 200 | 150 | 31,242 | ERR1760147 | A. halleri pop. AhaN4 |
We will download the files as follows:
srr.ids
1
2
3
4
ERS1475237
ERS1475238
ERS1475240
ERS1475241
To do so:
1
2
3
4
module load sratoolkit
module load parallel
parallel -a srr.ids prefetch --max-size 50GB
parallel -a srr.ids fastq-dump --split-files --origfmt --gzip
Since reference genome for this species of Arabidopsis is available, we will use it as reference. We will have to download the genome from the database
1
2
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-43/fasta/arabidopsis_halleri/dna/Arabidopsis_halleri.Ahal2.2.dna.toplevel.fa.gz
gunzip Arabidopsis_halleri.Ahal2.2.dna.toplevel.fa.gz
These datasets are all we need to get started. Although, the SRA download through prefetch is faster, it takes long time for converting sra file to fastq using fastq-dump. Alternatively, you can obtain and download fastq files directly form European Nucleotide Archive (ENA). The links are saved here if you want to use them instead (note the IDs are different, but they are from the same study and the results will be identical regardless of what data you use)
Organization
The files and folders will be organized as follows:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
├── 0_index
│ └── Arabidopsis_halleri.Ahal2.2.dna.toplevel.fa
├── 1_data
│ ├── ERR1760144_1.fastq
│ ├── ERR1760144_2.fastq
│ ├── ERR1760145_1.fastq
│ ├── ERR1760145_2.fastq
│ ├── ERR1760146_1.fastq
│ ├── ERR1760146_2.fastq
│ ├── ERR1760147_1.fastq
│ ├── ERR1760147_2.fastq
│ └── srr.ids
├── 2_fastqc
├── 3_pre-processing
├── 4_gatk-round-1
├── 5_recalibration
├── 6_gatk_round-2
└── 7_filtering
Workflow
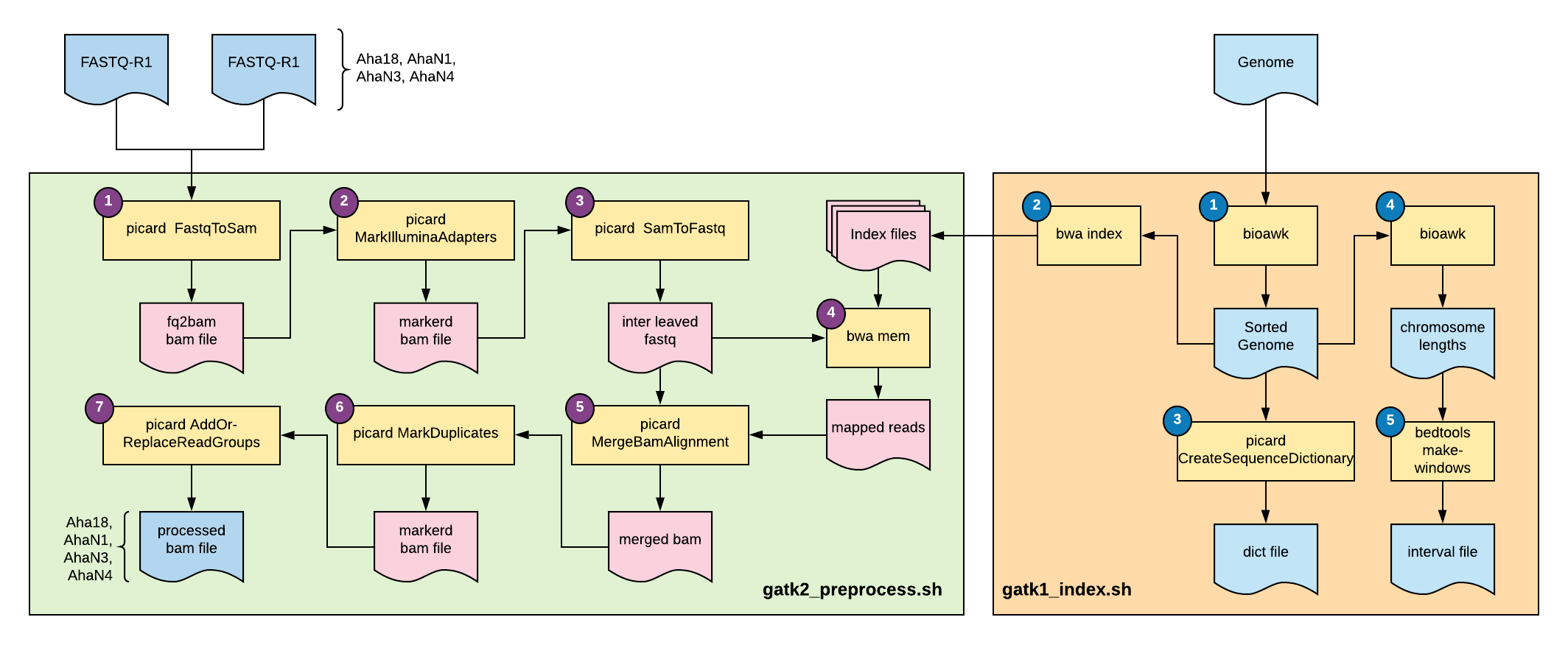
Fig 1: Overview of this tutorial
Step 0: Quality check the files
Soft link the fastq files and run FASTQC on them:
1
2
3
4
5
6
7
8
cd 2_fastqc
for fq in ../1_data/*.fastq; do
ln -s $fq
done
module load parallel
module load fastqc
parallel "fastqc {}" ::: *.fastq
cd ../
you can examine the results by opening each html page or you can merge them to a single report using multiqc. The data seems satisfactory, so we will proceed to next step.
Step 1: Getting the files ready for GATK
We will need the bwa index files and windows for processing small chunks of the genome in parallel.
gatk0_index.sh
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
cd 0_index
#!/bin/bash
# script to prepare reference genome for GATK snp calling
# indexes for BWA mapping porgram
# generates windows
# Arun Seetharam
# 5/16/2019
if [ $# -ne 2 ] ; then
echo -e "usage: $(basename "$0") <genome.fasta> <name>"
echo ""
echo -e "\tgenome.fasta:\tFASTA formatted reference genome"
echo -e "\tname:\tsmall name for easy reference management, any argument will suffice, must be one word"
echo ""
exit 0
fi
module load samtools
module load gatk
module load bwa
module load bedtools2
module load bioawk
ref="$1"
name="$2"
window=10000000
bioawk -c fastx '{print}' $ref | sort -k1,1V | awk '{print ">"$1;print $2}' | fold > ${name}.fasta
gatk CreateSequenceDictionary --REFERENCE ${name}.fasta --OUTPUT ${name}.dict
bwa index -a bwtsw ${name}.fasta
bioawk -c fastx '{print $name"\t"length($seq)}' ${name}.fasta > ${name}.length
bedtools makewindows -w $window -g ${name}.length |\
awk '{print $1"\t"$2+1"\t"$3}' |\
sed 's/\t/:/1' |\
sed 's/\t/-/1' > ${name}_coords.bed
run this as:
1
2
3
chmod +x gatk0_index.sh
./gatk0_index.sh Arabidopsis_halleri.Ahal2.2.dna.toplevel.fa ahalleri
cd ../
stdout for index step
``` 13:28:19.862 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/opt/rit/el9/20230413/app/linux-rhel9-x86_64_v3/gcc-11.2.1/gatk-4.3.0.0-qlpdiofa4jimy66vt2bn45pz3y5nmwea/bin/gatk-package-4.3.0.0-local.jar!/com/intel/gkl/native/libgkl_compression.so [Thu Nov 16 13:28:19 CST 2023] CreateSequenceDictionary OUTPUT=ahalleri.dict REFERENCE=ahalleri.fasta TRUNCATE_NAMES_AT_WHITESPACE=true NUM_SEQUENCES=2147483647 VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=2 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json USE_JDK_DEFLATER=false USE_JDK_INFLATER=false [Thu Nov 16 13:28:19 CST 2023] Executing as satheesh@nova18-5 on Linux 5.14.0-284.25.1.el9_2.x86_64 amd64; OpenJDK 64-Bit Server VM 11.0.17+8; Deflater: Intel; Inflater: Intel; Provider GCS is available; Picard version: 4.3.0.0 [Thu Nov 16 13:28:21 CST 2023] picard.sam.CreateSequenceDictionary done. Elapsed time: 0.03 minutes. Runtime.totalMemory()=2579496960 Tool returned: 0 [bwa_index] Pack FASTA... 1.18 sec [bwa_index] Construct BWT for the packed sequence... [BWTIncCreate] textLength=392486396, availableWord=39616672 [BWTIncConstructFromPacked] 10 iterations done. 65350012 characters processed. [BWTIncConstructFromPacked] 20 iterations done. 120730156 characters processed. [BWTIncConstructFromPacked] 30 iterations done. 169948156 characters processed. [BWTIncConstructFromPacked] 40 iterations done. 213689196 characters processed. [BWTIncConstructFromPacked] 50 iterations done. 252562300 characters processed. [BWTIncConstructFromPacked] 60 iterations done. 287108828 characters processed. [BWTIncConstructFromPacked] 70 iterations done. 317809836 characters processed. [BWTIncConstructFromPacked] 80 iterations done. 345092924 characters processed. [BWTIncConstructFromPacked] 90 iterations done. 369338156 characters processed. [BWTIncConstructFromPacked] 100 iterations done. 390883356 characters processed. [bwt_gen] Finished constructing BWT in 101 iterations. [bwa_index] 96.04 seconds elapse. [bwa_index] Update BWT... 0.73 sec [bwa_index] Pack forward-only FASTA... 0.77 sec [bwa_index] Construct SA from BWT and Occ... 43.17 sec [main] Version: 0.7.17-r1188 [main] CMD: bwa index -a bwtsw ahalleri.fasta [main] Real time: 147.423 sec; CPU: 141.898 sec ```Note: if you do not have bioawk module available on your HPC, you can install it in your home directory as follows:
1
2
3
4
5
6
7
8
ml bison
git clone https://github.com/lh3/bioawk.git
cd bioawk
make
mkdir -p ~/bin
mv maketab bioawk ~/bin/
echo 'export PATH=$PATH:~/bin' >> ~/.bashrc
source ~/.bashrc
Step 2: Preprocessing the fastq files
For handling purpose, we will rename the files to their respective population name and run it through the processing step that process the fastq files, algins and gets them read for variant calling.
id-names.txt
1
2
3
4
Aha18 ERR1760144
AhaN1 ERR1760145
AhaN3 ERR1760146
AhaN4 ERR1760147
1
2
3
4
while read a b; do
ln -s $(pwd)/1_data/${b}_1.fastq 3_pre-processing/${a}_R1.fastq
ln -s $(pwd)/1_data/${b}_2.fastq 3_pre-processing/${a}_R2.fastq
done<id-names.txt
generate commands and run the gatk2_preprocess.sh script
1
2
3
4
5
6
7
8
cd 3_pre-processing
for fq in *_R1.fastq; do
echo "./gatk2_preprocess.sh /work/LAS/mhufford-lab/arnstrm/ler/0_index/ahalleri.fasta $fq $(echo $fq |sed 's/_R1/_R2/g')"
done > process.cmds
./makeSLURMs.py 1 process.cmds
for sub in *.sub; do
sbatch $sub;
done
the script:
gatk2_preprocess.sh
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
#!/bin/bash
# script to prepare fastq files for GATK snp calling
# Arun Seetharam
# 5/16/2019
if [ $# -ne 3 ] ; then
echo -e "usage: $(basename "$0") <reference> <read R1> <read R2>"
echo ""
echo -e "\treference:\tindexed reference genome (full path)-short name in indexing script is fine"
echo -e "\tR1:\t forward read"
echo -e "\tR2:\t reverse read"
echo ""
exit 0
fi
module load bwa
module load samtools
ulimit -c unlimited
REF=$1
R1=$2
R2=$3
# adjust this to suit your input file name
OUT=$(echo $R1 |cut -f 1-3 -d "_")
JAVA_OPTIONS="-Xmx100g -Djava.io.tmpdir=$TMPDIR"
# platform id from fastq file
if [ ${R1: -3} == ".gz" ]; then
PLT=$(zcat $R1 |head -n 1 |cut -f 3 -d ":")
RGPU=$(zcat $R1 |head -n 1 |cut -f 3-5 -d ":")
else
PLT=$(cat $R1 |head -n 1 |cut -f 3 -d ":")
RGPU=$(cat $R1 |head -n 1 |cut -f 3-5 -d ":")
fi
# time stamp as string of numbers
TDATE=$(date '+%Y-%m-%d %H:%M:%S' |sed 's/ /T/g')
# read group identifier, should be unique, usually genotype name
RGID=$(echo $R1 |cut -f 1-3 -d "_")
# library identifier
RGLB="$RGID"
# platform name choose either ILLUMINA, SOLID, LS454, HELICOS and PACBIO
RGPL="ILLUMINA"
# genotype name, this will appear in VCF file header
RGSM="$RGID"
# convert fastq to sam and add readgroups
gatk --java-options ${JAVA_OPTIONS} FastqToSam \
--FASTQ ${R1} \
--FASTQ2 ${R2} \
--OUTPUT ${OUT}_fastqtosam.bam \
--READ_GROUP_NAME ${OUT} \
--SAMPLE_NAME ${OUT}_name \
--LIBRARY_NAME ${OUT}_lib \
--PLATFORM_UNIT ${PLT} \
--PLATFORM illumina \
--SEQUENCING_CENTER ISU \
--RUN_DATE ${TDATE} || {
echo >&2 ERROR: FastqToSam failed for $OUT
exit 1
}
# marking adapters
gatk --java-options ${JAVA_OPTIONS} MarkIlluminaAdapters \
--INPUT ${OUT}_fastqtosam.bam \
--OUTPUT ${OUT}_markilluminaadapters.bam \
--METRICS ${OUT}_markilluminaadapters_metrics.txt \
--TMP_DIR ${TMPDIR} || {
echo >&2 ERROR: MarkIlluminaAdapters failed for $OUT
exit 1
}
# convert bam back to fastq for mapping
gatk --java-options ${JAVA_OPTIONS} SamToFastq \
--INPUT ${OUT}_markilluminaadapters.bam \
--FASTQ ${OUT}_samtofastq_interleaved.fq \
--CLIPPING_ATTRIBUTE XT \
--CLIPPING_ACTION 2 \
--INTERLEAVE true \
--INCLUDE_NON_PF_READS true \
--TMP_DIR ${TMPDIR} || {
echo >&2 ERROR: SamToFastq failed for $OUT
exit 1
}
# mapping reads to indexed genome
bwa mem \
-M \
-t 15 \
-p $REF \
${OUT}_samtofastq_interleaved.fq |\
samtools view -buS - > ${OUT}_bwa_mem.bam || {
echo >&2 ERROR: BWA failed for $OUT
exit 1
}
# merging alignments
gatk --java-options ${JAVA_OPTIONS} MergeBamAlignment \
--REFERENCE $REF \
--UNMAPPED_BAM ${OUT}_fastqtosam.bam \
--ALIGNED_BAM ${OUT}_bwa_mem.bam \
--OUTPUT ${OUT}_snippet_mergebamalignment.bam \
--CREATE_INDEX true \
--ADD_MATE_CIGAR true --CLIP_ADAPTERS false \
--CLIP_OVERLAPPING_READS true \
--INCLUDE_SECONDARY_ALIGNMENTS true \
--MAX_INSERTIONS_OR_DELETIONS -1 \
--PRIMARY_ALIGNMENT_STRATEGY MostDistant \
--ATTRIBUTES_TO_RETAIN XS \
--TMP_DIR "${TMPDIR}" || {
echo >&2 ERROR: MergeBamAlignment failed for $OUT
exit 1
}
# mark duplicates
gatk --java-options ${JAVA_OPTIONS} MarkDuplicates \
--INPUT ${OUT}_snippet_mergebamalignment.bam \
--OUTPUT ${OUT}_prefinal.bam \
--METRICS_FILE ${OUT}_mergebamalignment_markduplicates_metrics.txt \
--OPTICAL_DUPLICATE_PIXEL_DISTANCE 2500 \
--CREATE_INDEX true \
--TMP_DIR $TMPDIR || {
echo >&2 ERROR: MarkDuplicates failed for $OUT
exit 1
}
# add read groups
gatk --java-options ${JAVA_OPTIONS} AddOrReplaceReadGroups \
--INPUT ${OUT}_prefinal.bam \
--OUTPUT ${OUT}_final.bam \
--RGID ${RGID} \
--RGLB ${RGLB} \
--RGPL ${RGPL} \
--RGPU ${RGPU} \
--RGSM ${RGSM} \
--CREATE_INDEX true \
--TMP_DIR $TMPDIR || {
echo >&2 ERROR: Adding read groups failed for $OUT
exit 1
}
echo >&2 "ALL DONE!"
# cleanup
rm ${OUT}_fastqtosam.bam
rm ${OUT}_markilluminaadapters.bam
rm ${OUT}_samtofastq_interleaved.fq
rm ${OUT}_bwa_mem.bam
rm ${OUT}_snippet_mergebamalignment.bam
rm ${OUT}_snippet_mergebamalignment.bai
At the end of this step, you will have the following files as output:
1
2
3
4
Aha18_final.bam
AhaN1_final.bam
AhaN4_final.bam
AhaN3_final.bam
Step 3: GATK round 1 variant calling
At this step, you will need the indexed genome and interval list (coords.bed) from the Step 0. Running the script will generate the commands that you will need to submit as slurm script as before.
Before starting, setup the files/folders as follows:
1
2
3
4
cd 4_gatk-round-1
for finalbam in ../3_pre-processing/*_final.ba?; do
ln -s $finalbam
done
The gatk3_cmdsgen.sh is as follows:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
#!/bin/bash
# script to generate GATK commands for snp calling
# gatk haplotype caller
# for each windows specified
# Arun Seetharam
# 5/16/2019
if [ $# -lt 3 ] ; then
echo -e "usage: $(basename "$0") <windows bed file> <genome fasta file> <regex for bam files>"
echo ""
echo -e "\twindows.bed:\tBED formatted reference intervals to call SNPs"
echo -e "\tgenome.fasta:\tFASTA formatted reference genome"
echo -e "\tfinal.bam files:\tBAM formatted final files from processing step"
echo ""
exit 0
fi
unset -v bamfiles
list="$1"
bamfiles=(*"${3}")
REF="$3"
for bam in ${bamfiles[@]}; do
echo -en "-I ${bam} ";
done > CombinedBAM_temp
while read line; do
if ! [[ $line == @* ]]; then
g2=$(echo $line | awk '{print $1":"$2"-"$3}'); \
g1=$(echo $line | awk '{print $1"_"$2"_"$3}'); \
CWD=$(pwd)
echo "gatk --java-options \"-Xmx80g -XX:+UseParallelGC\" HaplotypeCaller -R ${REF} $(cat CombinedBAM_temp) -L "${g2}" --output "${g1}".vcf;";
fi
done<${list}
Make the script executable.
1
chmod +x gatk3_cmdsgen.sh
Run this script as:
1
./gatk3_cmdsgen.sh ../0_index/ahalleri_coords.bed ../0_index/ahalleri.fasta *final.bam > gatk.cmds
This will generate 2239 commands (one gatk command per interval). Since the GATK 4 cannot use multiple threads, you can run one job per thread and thus fit multiple jobs in a single node. Using multiple nodes, you can run these commands much faster than running a single command on a bigger interval or a whole genome.
Before, you ran makeSLURMs.py script. This job runs the commands serially. Another script makeSLURMp.py also does the same thing, but instead it runs the command in parallel. We will use that and specify how many jobs we want to run. To split them
1
2
3
4
5
6
7
../3_pre-processing/makeSLURMp.py 220 gatk.cmds
# some fixing is needed to make sure that it runs the right number of jobs
# and then submit
for sub in *.sub; do
sed -i 's/parallel -j 1 --joblog/parallel -j 18 --joblog/g' $sub;
sbatch $sub;
done
This will run 18 jobs at time and 220 jobs total, per node. Upon completion, you will see many VCF file (2239 total) and its associated index files (idx)
Next step is to merge and perform filtering on these variants to use them to re-calibrate the bam files. The re-calibrated bam files will be then used for calling variants in the similar fashion.
Run the gatk4_filter.sh for merging, filtering and cleaning-up files
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
#!/bin/bash
# script to filter snps
# gatk tutorial
# Arun Seetharam
# 5/16/2019
merged=merged
#change name as you wish
ref=$1
if [ $# -lt 1 ] ; then
echo -e "usage: $(basename "$0") <genome fasta file>"
echo ""
echo -e "\tgenome.fasta:\tFASTA formatted reference genome"
echo ""
exit 0
fi
module load vcftools
module load GIF/datamash
module load gatk
module load bcftools
mkdir -p vcffiles idxfiles
# merge vcf files
mv *.vcf ./vcffiles
mv *.idx ./idxfiles
cd vcffiles
vcf=(*.vcf)
module load parallel
parallel "grep -v '^#' {}" ::: *.vcf >> ../${merged}.body
grep "^#" ${vcf[1]} > ../${merged}.head
cd ..
cat ${merged}.head ${merged}.body >> ${merged}.vcf
cat ${merged}.vcf | vcf-sort -t $TMPDIR -p 36 -c > ${merged}_sorted.vcf
# calculate stats
bcftools stats ${merged}_sorted.vcf > ${merged}_sorted.vchk
plot-vcfstats ${merged}_sorted.vchk -p plots/
maxdepth=$(grep -oh ";DP=.*;" ${merged}_sorted.vcf | cut -d ";" -f 2 | cut -d "=" -f 2 | datamash mean 1 sstdev 1 | awk '{print $1+$2*5}')
# separate SNPs and INDELs
vcftools --vcf ${merged}_sorted.vcf --keep-only-indels --recode --recode-INFO-all --out ${merged}_sorted-indels.vcf
vcftools --vcf ${merged}_sorted.vcf --remove-indels --recode --recode-INFO-all --out ${merged}_sorted-snps.vcf
gatk --java-options \"-Xmx80g -XX:+UseParallelGC\" VariantFiltration \
--reference $ref \
--variant ${merged}_sorted-snps.vcf \
--filter-expression "QD < 2.0 || FS > 60.0 || MQ < 45.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0 || DP > ${maxdepth}" \
--filter-name "FAIL" \
--output ${merged}_filtered-snps.vcf
gatk --java-options \"-Xmx80g -XX:+UseParallelGC\" VariantFiltration \
--reference $ref \
--variant ${merged}_sorted-indels.vcf \
--filter-expression "QD < 2.0 || FS > 200.0 || ReadPosRankSum < -20.0" \
--filter-name "FAIL" \
--output ${merged}_filtered-indels.vcf
Run this script as:
1
2
cd 4_gatk-round-1
gatk4_filter.sh ../0_index/ahalleri.fasta
After completion, you will have the first round results for SNP calling. Technically, this can be used as results, the best practices recommend that you run another round of SNP calling using this results to calibrate the original BAM files.
The main results from this script are:
1
2
merged_filtered-snps.vcf
merged_filtered-indels.vcf
Step 4: Variant Recalibration
As mentioned before, we will run the recalibration of each BAM file with the script gatk5_bsqr.sh in 5_recalibration folder.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
#!/bin/bash
# script to bsqr for SNP calling
# gatk SNP calling tutorial
# Arun Seetharam
# 5/16/2019
if [ $# -lt 3 ] ; then
echo -e "usage: $(basename "$0") <genome file> <VCF file> <bam file>"
echo ""
echo -e "\tgenome.fasta:\tFASTA formatted reference genome"
echo -e "\tvcffile:\tVCF format first round filtered SNPs, you can also use known SNPs from other sources as well"
echo -e "\tfinal.bam files:\tBAM formatted final files from processing step"
echo ""
exit 0
fi
module load gatk
module load r-geneplotter
REF="$1"
FRVCF="$2"
IBAM="$3"
OBAM=$(basename ${IBAM} | sed 's/_final.bam//g')
gatk BaseRecalibrator \
--reference $REF \
--input $IBAM \
--known-sites ${FRVCF} \
--output ${OBAM}_bef-R1.table
gatk ApplyBQSR \
--reference $REF \
--input $IBAM \
--output ${OBAM}_recal.bam \
--bqsr-recal-file ${OBAM}_bef-R1.table
gatk BaseRecalibrator \
--reference $REF \
--input ${OBAM}_recal.bam \
--known-sites ${FRVCF} \
--output ${OBAM}_aft-R1.table
gatk AnalyzeCovariates \
-before ${OBAM}_bef-R1.table \
-after ${OBAM}_aft-R1.table \
-plots ${OBAM}-AnalyzeCovariates.pdf
Now, create commands and slurm submission script to run them on each BAM file
1
2
3
4
5
6
7
8
9
10
11
cd 5_recalibration
for bam in ../3_pre-processing/*final.bam; do
ln -s $bam
done
for bam in *_final.bam; do
echo "./GATK_06_BSQR.sh $bam"
done > bsqr.cmds
makeSLURMs.py 1 bsqr.cmds
for sub in bsqr_?.sub; do
sbatch $sub;
done
upon completion, you will have BAM files with _recal.bam suffix. You will need to rerun the entire process of step 3 using these files.
Step 5: GATK round 2 variant calling
We will now move to folder 6_gatk_round-2 and re-run the GATK SNPcalling. You can easily reuse all the SLURM scripts that you generated in the step3
Organize:
1
2
3
4
cd 6_gatk_round-2
for recalbam in ../5_recalibration/*_recal.ba?; do
ln -s $recalbam
done
Next, run the commands generator script:
1
gatk3_cmdsgen.sh ../0_index/ahalleri_coords.bed ../0_index/ahalleri.fasta *recal.bam > gatk.cmds
This will generate 2239 commands (one gatk command per interval).
1
2
3
4
5
6
7
makeSLURMp.py 220 gatk.cmds
# some fixing is needed to make sure that it runs the right number of jobs
# and then submit
for sub in *.sub; do
sed -i 's/parallel -j 1 --joblog/parallel -j 18 --joblog/g' $sub;
sbatch $sub;
done
Once all the jobs complete, run filtering script:
1
2
cd 6_gatk_round-2
gatk4_filter.sh ../0_index/ahalleri.fasta
The will create final results for SNP calling:
1
2
merged_filtered-snps.vcf
merged_filtered-indels.vcf