
Have you ever wanted to figure out which reads are the most useful for creating a more contiguous assembly? Here I assemble a nematode genome, assess contamination using Blobtools2, extract non-contaminant reads, and reassemble.
Dependencies
- FLYE – assembler
- BLAST-plus – for blasting to NCBI nt for blobtools
- Minimap2 – read mapping for blobtools
- Samtools – extracting the reads
- Bioawk – creating bed files to extract reads
- Cdbfasta – used to extract non-contaminant contigs
- new_Assemblathon.pl – to assess assembly quality – https://github.com/ISUgenomics/common_scripts/blob/master/new_Assemblathon.pl
First assembly
1
2
3
4
/work/gif/remkv6/USDA/11_Experiment/03_MysteryFlye
ln -s ../MysteryReadsAllNanopore.fastq.gz
ml miniconda3; source activate flye;flye --nano-raw MysteryReadsAllNanopore.fastq.gz -g 140m -o /work/gif/remkv6/USDA/11_Experiment/03_MysteryFlye -t 36
Check initial assembly quality
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
~/common_scripts/new_Assemblathon.pl assembly.fasta
---------------- Information for assembly 'assembly.fasta' ---------------
Number of scaffolds 5222
Total size of scaffolds 204772733
Longest scaffold 751399
Shortest scaffold 68
Number of scaffolds > 1K nt 4700 90.0%
Number of scaffolds > 10K nt 3134 60.0%
Number of scaffolds > 100K nt 499 9.6%
Number of scaffolds > 1M nt 0 0.0%
Number of scaffolds > 10M nt 0 0.0%
Mean scaffold size 39213
Median scaffold size 17620
N50 scaffold length 90687
L50 scaffold count 584
n90 scaffold length 23396
L90 scaffold count 2277
scaffold %A 29.54
scaffold %C 20.45
scaffold %G 20.45
scaffold %T 29.55
scaffold %N 0.00
scaffold %non-ACGTN 0.00
Number of scaffold non-ACGTN nt 0
Identify contamination in this initial assembly with Blobtools2
map nanopore reads to assembly
This can be done with any reads, though mapping long reads with minimap2 is ridiculously fast.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
/work/gif/remkv6/USDA/11_Experiment/06_BlobtoolsDrafts/01_Mystery/01_nanoporeMapping
ln -s ../../../03_MysteryFlye/MysteryReadsAllNanopore.fastq.gz
ln -s ../../../03_MysteryFlye/assembly.fasta
# USAGE sh runMinimap.sh query.fasta target.fasta
#############################################################################################
#!/bin/bash
query=$1
target=$2
outname="${query%.*}_${target%.*}_minimap2.bam"
module load minimap2
minimap2 -x map-ont -k15 -a -t 36 $target $query > ${outname}
mkdir ${query%.*}dir; ml samtools
samtools view --threads 24 -b -o ${outname%.*}.bam ${outname}
samtools sort -o ${outname%.*}_sorted.bam -T ${query%.*}dir --threads 24 ${outname%.*}.bam
#############################################################################################
sh runMinimap.sh MysteryReadsAllNanopore.fastq.gz assembly.fasta
Checking my mapping percentages to see if they are reasonable
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
ml samtools; samtools flagstat
2129548 + 0 in total (QC-passed reads + QC-failed reads)
656411 + 0 secondary
337474 + 0 supplementary
0 + 0 duplicates
1612546 + 0 mapped (75.72% : N/A)
0 + 0 paired in sequencing
0 + 0 read1
0 + 0 read2
0 + 0 properly paired (N/A : N/A)
0 + 0 with itself and mate mapped
0 + 0 singletons (N/A : N/A)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
Run a megablast on your assembly
Note, if you do not have -task megablast set, the blast will take much longer. Also note the -outfmt, it must be in this format to attribute taxonomy info for blobtools
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
/work/gif/remkv6/USDA/11_Experiment/06_BlobtoolsDrafts/01_Mystery/02_megablast
#softlink genome
ln -s ../../../05_juicer/01_Mystery/references/MysteryGenome.fasta
#run the script below
sh runMegablast.sh MysteryGenome.fasta
#!/bin/bash
#runMegablast.sh
#Created by Rick Masonbrink 08/11/21
#usage
#sh runMegablast.sh genome.fasta
################################################################################
wget ftp://ftp.ncbi.nlm.nih.gov/blast/db/taxdb.tar.gz
tar -zxvf taxdb.tar.gz
ml gcc/10.2.0-zuvaafu; ml blast-plus/2.11.0-py3-4pqzweg
FASTA="$1"
blastn \
-query ${FASTA} \
-task megablast \
-db /work/gif/databases/Blast/NT-DB/nt \
-outfmt '6 qseqid staxids bitscore std sscinames sskingdoms stitle' \
-culling_limit 10 \
-num_threads 36 \
-evalue 1e-3 \
-out ${FASTA%.**}.vs.nt.cul5.1e3.megablast.out
################################################################################
Run blobtools2
Set up file structure for blobtools
1
2
3
/work/gif/remkv6/USDA/11_Experiment/06_BlobtoolsDrafts/01_Mystery/03_blobtools
ln -s ../01_nanoporeMapping/MysteryReadsAllNanopore_sorted.bam
ln -s ../02_megablast/MysteryGenome.vs.nt.cul5.1e3.megablast.out
Get the taxon info and move it into a folder
1
2
3
4
wget https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/new_taxdump/new_taxdump.tar.gz
tar -zxvf new_taxdump.tar.gz
mkdir taxdump
mv *.dmp taxdump/.
Execute Blobtools 2 – must be on a cpu node that can connect to the internet
1
2
3
ml singularity; ml blobtools2
singularity shell /opt/rit/singularity/images/blobtools2/2.2.0/blobtools2.simg
blobtools create --fasta assembly.fasta --cov MysteryReadsAllNanopore_sorted.bam --hits MysteryGenome.vs.nt.cul5.1e3.megablast.out --replace --taxdump taxdump Mystery
Once done, connect to novaDTN (internet capable node) through your personal computers terminal
1
2
3
4
5
6
7
8
9
10
11
12
ssh -L 8001:127.0.0.1:8001 -L 8000:127.0.0.1:8000 remkv6@novadtn.its.iastate.edu
#Navigate the directory of your blobtools run
/work/gif/remkv6/USDA/11_Experiment/06_BlobtoolsDrafts/01_Mystery/03_blobtools
ml singularity; ml blobtools2
singularity shell /opt/rit/singularity/images/blobtools2/2.2.0/blobtools2.simg
#this freqently fails, but must go through channel 8001 to work. Must get out of singularity and back in to reset it.
blobtools view --interactive Mystery
#then paste the link into your personal computer's browser
BlobPlotCircle
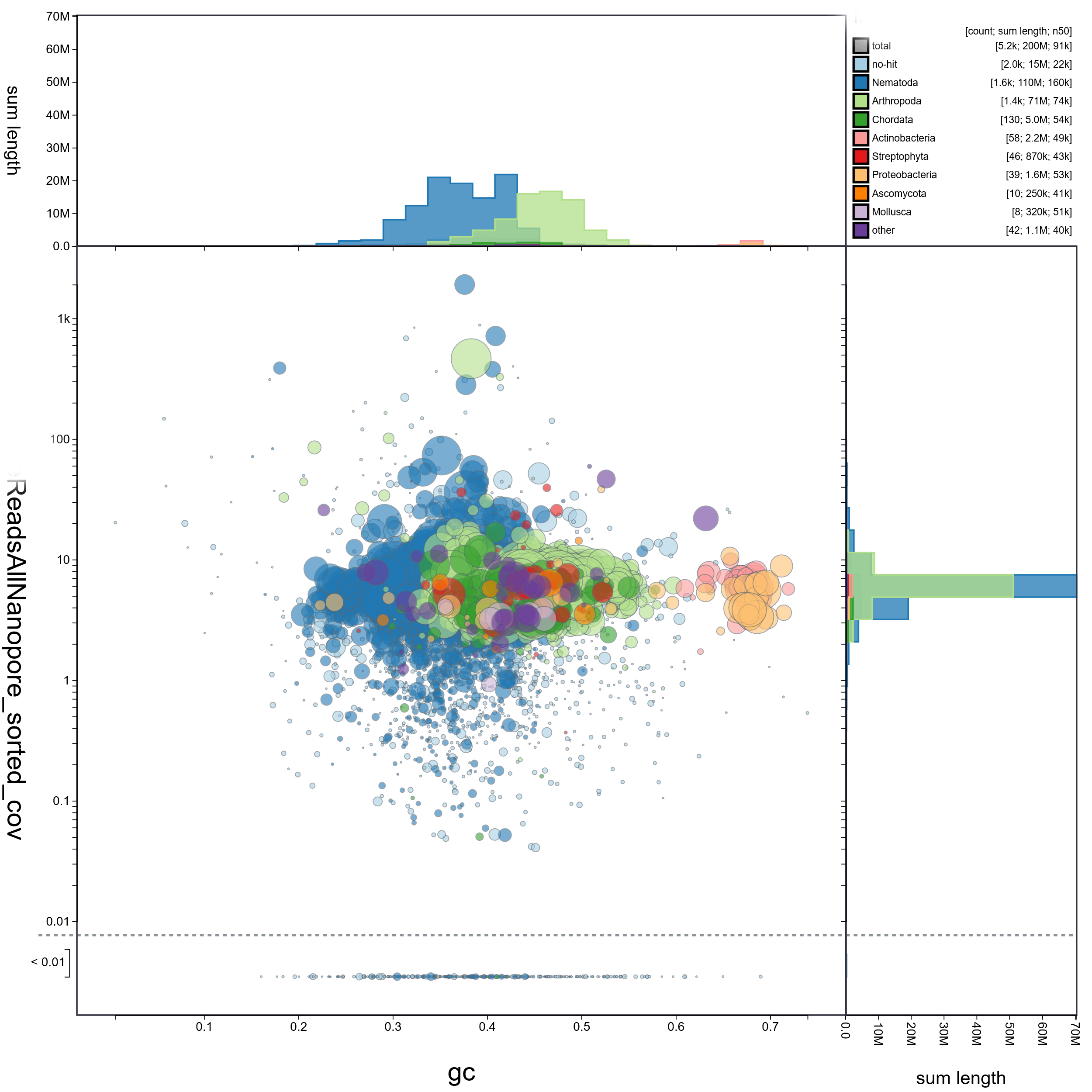
BLOBTABLE.CSV
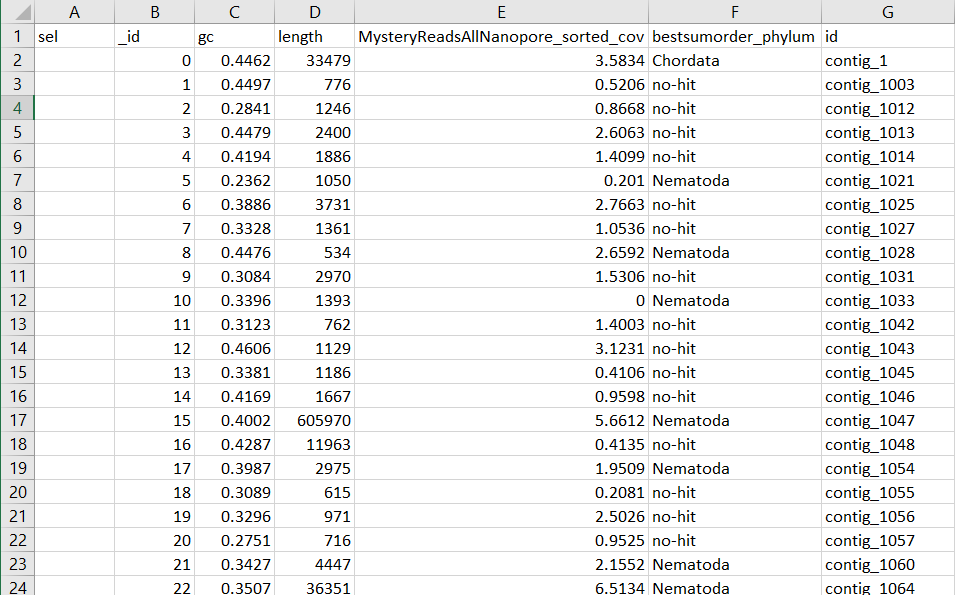
Yay, you now can filter contigs that are contaminants. The only file needed is the blobtools.csv output, though I grab the circle blob plot also. With my nematode assembly, I frequently get contigs attributed to arthropoda, as they more highly represented sequences in NT.
Filter the assembly using blobtools output
I have lots of real contaminants in my assembly, so I am hard-filtering. Contigs/scaffolds had to be attributed to Nematoda, Arthropoda, or no-hit, and no-hit must have coverage.
1
less MysteryBlobTable.csv |sed 's/,/\t/g' |sed 's/"//g' |cut -f 2- |awk '$5=="Nematoda" || $5=="no-hit" || $5=="Arthropoda"' |awk '{if($5=="no-hit" && $4==0) {next} else {print $0}} ' |awk '$3>1999 {print $6}' |cdbyank assembly.fasta.cidx >FilteredMysteryGenome.fasta
Make sure you arent missing contigs that were mislabeled as other taxa by blobtools, by searching for your most closely related species in your blast output. I did not have any mislabeled contigs, so nothing was added here.
1
less MysteryBlobTable.csv |sed 's/,/\t/g' |sed 's/"//g' |cut -f 2- |awk '$5!="Nematoda" && $5!="no-hit" && $5!="Arthropoda"' |awk '$3>10000 {print $6}' |grep -w -f - MysteryGenome.vs.nt.cul5.1e3.megablast.out |grep "Heterodera glycines" - |awk '{print $1}' |sort|uniq|cdbyank MysteryGenome.fasta.cidx >>FilteredMysteryGenome.fasta
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
~/common_scripts/new_Assemblathon.pl FilteredMysteryGenome.fasta
---------------- Information for assembly 'FilteredMysteryGenome.fasta' ----------------
Number of scaffolds 3865
Total size of scaffolds 192263859
Longest scaffold 751399
Shortest scaffold 2001
Number of scaffolds > 1K nt 3865 100.0%
Number of scaffolds > 10K nt 2871 74.3%
Number of scaffolds > 100K nt 489 12.7%
Number of scaffolds > 1M nt 0 0.0%
Number of scaffolds > 10M nt 0 0.0%
Mean scaffold size 49745
Median scaffold size 27137
N50 scaffold length 95784
L50 scaffold count 520
n90 scaffold length 24803
L90 scaffold count 2027
scaffold %A 29.81
scaffold %C 20.18
scaffold %G 20.19
scaffold %T 29.82
scaffold %N 0.00
scaffold %non-ACGTN 0.00
Number of scaffold non-ACGTN nt 0
Extract only reads that do not map to contaminant contigs
The read name and orientation of the contaminant reads, are extracted with the fastq if you do not do this step.
1
bioawk -c fastx '{print $name,"1",length($seq)}' FilteredMysteryGenome.fasta|tr " " "\t" >KeeperContigs.bed
Extract the reads mapping to the contigs you want to keep, and convert to fastq
1
2
samtools view -b -L KeeperContigs.bed -o CleanedSCNReads.bam ../01_nanoporeMapping/MysteryReadsAllNanopore.bam
samtools fastq CleanedSCNReads.bam >CleanedSCNReads.fastq
Filter fastq by length of at least 2kb
1
bioawk -cfastx 'length($seq)>=1999{print "@"$name"\n"$seq"\n+\n"$qual}' CleanedSCNReads.fastq >Long2kCleanedSCNReads.fastq
Assembly 2 without any contaminating reads or reads shorter than 2kb
1
2
3
ln -s ../06_BlobtoolsDrafts/01_Mystery/03_blobtools/Long5kCleanedSCNReads.fastq
ml miniconda3; source activate flye;flye --nano-raw Long2kCleanedSCNReads.fastq -g 140m -o /work/gif/remkv6/USDA/11_Experiment/03_MysteryFlye -t 36
Check assembly quality and gaze in awe at the improved contiguity
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
~/common_scripts/new_Assemblathon.pl
Number of scaffolds 2201
Total size of scaffolds 155445271
Longest scaffold 1053850
Shortest scaffold 431
Number of scaffolds > 1K nt 2138 97.1%
Number of scaffolds > 10K nt 1753 79.6%
Number of scaffolds > 100K nt 468 21.3%
Number of scaffolds > 1M nt 1 0.0%
Number of scaffolds > 10M nt 0 0.0%
Mean scaffold size 70625
Median scaffold size 43946
N50 scaffold length 131176
L50 scaffold count 321
n90 scaffold length 40032
L90 scaffold count 1179
scaffold %A 30.17
scaffold %C 19.80
scaffold %G 19.80
scaffold %T 30.23
scaffold %N 0.00
scaffold %non-ACGTN 0.00
Number of scaffold non-ACGTN nt 0