
Introduction
Genomic structural variation plays a major role in genome evolution and function, influencing genetic diversity, disease susceptibility, and adaptation. Among this variation, transposable elements (TEs) are particularly intriguing due to their ability to transpose and duplicate within a genome, potentially disrupting genes or regulatory regions. This tutorial provides a step-by-step guide to assessing structural variation in your genome using SVIM and identifying transposons that have recently jumped. You will learn how to:
- Prepare input data for SVIM analysis.
- Map long reads to your genome.
- Run SVIM to detect structural variants.
- Compare SVIM output with repeat annotations to identify TE activity.
Prerequisites
Data Requirements
- An annotated genome
- Long read sequencing data
- A repeat annotation for your genome
Software
- minimap2 for read mapping
- samtools for sorting and indexing BAM files
- bedtools for comparisons of genomic coordinates
- tabix for visualization
Installation
1
2
3
/work/gif/remkv6/USDA/04_TEJumper
conda create -n svim_env --channel bioconda svim
source activate svim_env
Mapping Long Reads to the Genome
Before mapping, create the temp folders for samtools
1
for f in *fastq; do mkdir ${f%.*}tmp; done
Mapping Reads with minimap2 and processing with samtools The following command maps reads to the genome, converts the files to bam, and indexes them.
1
minimap2 -a -x map-ont MindFlayergenome.fasta EvilPowers_1_1fastq/EvilPowers1_1.fastq | samtools sort -T EvilPowers_1_1fastq/EvilPowers1_1tmp -o EvilPowers_1_1fastq/EvilPowers1_1.sorted.bam; samtools index EvilPowers_1_1fastq/EvilPowers1_1.sorted.bam
Run SVIM on your mapped reads
Navigate to your working directory
1
/work/gif/remkv6/USDA/04_TEJumper
Run SVIM
1
for f in *bam; do echo "ml miniconda3; source activate svim_env; svim alignment /work/gif/remkv6/USDA/04_Pseudogenome/ "$f" MindFlayergenome.fasta";done >svim.sh
svim.sh -- Click to see content
ml miniconda3; source activate svim_env; svim alignment /work/gif/remkv6/USDA/04_Pseudogenome/ EvilPowers1_1.sorted.bam MindFlayergenome.fasta ml miniconda3; source activate svim_env; svim alignment /work/gif/remkv6/USDA/04_Pseudogenome/ EvilPowers1_2.sorted.bam MindFlayergenome.fasta ml miniconda3; source activate svim_env; svim alignment /work/gif/remkv6/USDA/04_Pseudogenome/ EvilPowers2_1.sorted.bam MindFlayergenome.fasta ml miniconda3; source activate svim_env; svim alignment /work/gif/remkv6/USDA/04_Pseudogenome/ EvilPowers2_2.sorted.bam MindFlayergenome.fasta
Understanding SVIM output
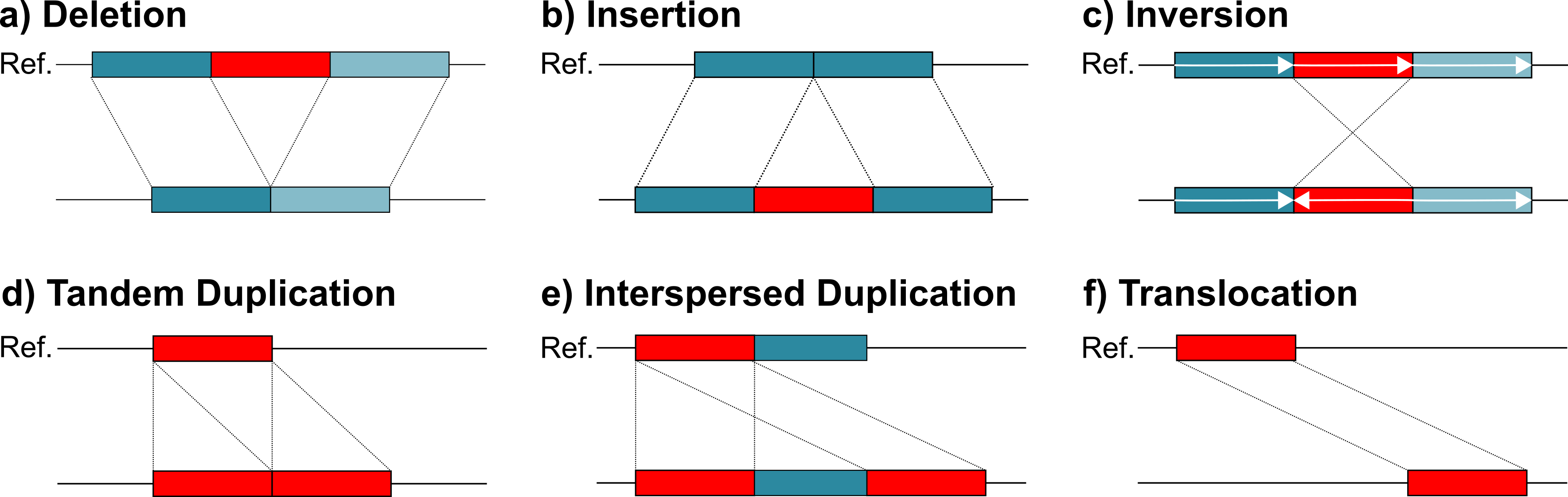
This is a fantastic illustration of SVIM output source. Within your candidates/ and signatures/ folders you will have bed files corresponding to each of these different types. While transposons may be associated with insertions and deletions, the other four categories can provide a clearer picture of genome source and destination for these jumping genes.
Identifying TE Activity
Here we will compare SVIM output with the genomic repeat annotation to build TE jumping candidates
Move to output directory (signatures have a higher confidence than the candidates)
1
/work/gif/remkv6/USDA/04_TEJumper/signatures
Softlink the repeat annotation. I used an existing RepeatModeler/RepeatMasker gff3.
1
ln -s /work/gif/remkv6/04_DovetailMindFlayerGenome/49_RenameChromosomes/01_Transfer2Box/RepeatMaskerFormatted.gff3
Identify repeats that overlap with these structural variants. Unless your genome is a model, this takes a little guesswork and manual assessment of duplication associations with TEs. Start by taking a look at overlaps between your SVIM bed files and your transposon annotations. Ideally you are looking for a complete duplication source and destination overlap with the same type of repeat. This doesnt happen very often with this dataset, so I concatenated all of the SVIM bed files.
Here use bedtools to find overlaps between structural variants and transposons.
1
2
ml bedtools2
bedtools intersect -wo -f .2 -a dup_int.bed -b RepeatMaskerFormatted.gff3 |less -S
In my dataset, TE jumps were not that plentiful, so I concatenated all bed files in attempt to find the goal I mention above (complete duplication source and destination overlap). Here, I remove variant events that are associated with supplemental alignments and remove most of the short structural variants to limit the data noise.
Filter the bed files for size of duplication and remove supplemental alignments.
1
cat *bed |grep -v "suppl" |awk '($3-$2)>1000' >LargeEverythingNoSuppl.bed
Examine variant and repeat overlaps with at least 20% overlap. Variants only from non-supplemental alignments and variants above 1kb.
1
bedtools intersect -wo -f .2 -a LargeEverythingNoSuppl.bed -b RepeatMaskerFormatted.gff3|less -S
What is my most actively modified transposon/repeat?
To determine which transposon family exhibits the most structural modifications:
1
2
3
4
5
6
7
8
9
10
11
ml bedtools2
bedtools intersect -wo -f .2 -a LargeEverythingNoSuppl.bed -b RepeatMaskerFormatted.gff3 \
| sort -k1,1V -k2,3nr \
| cut -f 15 \
| sed 's/:/\t/g' \
| sed 's/\.\./\t/g' \
| cut -f 2 \
| sort \
| uniq -c \
| sort -k1,1nr \
| less
Results of above command -- Click to see content
18 rnd-5_family-3824 14 rnd-3_family-296 12 rnd-4_family-1175 12 rnd-4_family-554 10 rnd-4_family-3941 10 rnd-4_family-586 10 rnd-5_family-506 8 rnd-3_family-742 8 rnd-4_family-1889 8 rnd-4_family-233 8 rnd-4_family-3318 8 rnd-4_family-871 8 rnd-5_family-2024 8 rnd-5_family-62 6 rnd-3_family-259 6 rnd-3_family-755 6 rnd-4_family-1030 6 rnd-4_family-1939 6 rnd-4_family-2286 6 rnd-4_family-615 6 rnd-4_family-636 6 rnd-4_family-676 6 rnd-4_family-76 6 rnd-4_family-78 6 rnd-5_family-1296 6 rnd-5_family-2138 6 rnd-5_family-2592 6 rnd-5_family-2847 6 rnd-5_family-3299 6 rnd-5_family-3786 6 rnd-5_family-884 4 rnd-3_family-136 4 rnd-3_family-45 4 rnd-3_family-675 4 rnd-3_family-92 4 rnd-4_family-1093 4 rnd-4_family-1163 4 rnd-4_family-136 4 rnd-4_family-1655 4 rnd-4_family-2265 4 rnd-4_family-2283 4 rnd-4_family-2901 4 rnd-4_family-294 4 rnd-4_family-3130 4 rnd-4_family-329 4 rnd-4_family-344 4 rnd-4_family-36 4 rnd-4_family-397
Since rnd-5_family-3824 is the most abundantly modified, lets look at all possible modified areas with this repeat.
1
2
3
4
5
bedtools intersect -wo -f .2 -a LargeEverythingNoSuppl.bed -b RepeatMaskerFormatted.gff3 \
| grep "rnd-5_family-3824" \
| sort \
| uniq \
| less -S
Results of above command -- Click to see content
Chromosome_1 21394028 21395344 INS;1;None;None 1.0 [Chromosome_1|21394028|21395344|INS;cigar|afa26991-52ed-4c97-964a-ed17aace0ded] Chromosome_1 RepeatMasker similarity 21394241 21394627 3.7 - . ID=Motif:rnd-5_family-3824..1769-2162#166139 387 Chromosome_1 21394028 21395344 INS;1;None;None 1.0 [Chromosome_1|21394028|21395344|INS;cigar|afa26991-52ed-4c97-964a-ed17aace0ded] Chromosome_1 RepeatMasker similarity 21395018 21395525 0.8 - . ID=Motif:rnd-5_family-3824..597-1126#166142 327 Chromosome_3 11203022 11204503 INS;1;None;None 1.0 [Chromosome_3|11203022|11204503|INS;cigar|311e9af7-3a96-41a9-a805-dd0ae1ba4684] Chromosome_3 RepeatMasker similarity 11203770 11204822 1.9 - . ID=Motif:rnd-5_family-3824..613-1659#80436 734 Chromosome_3 13650014 13651306 DEL;1;None;None 1.0 [Chromosome_3|13650014|13651306|DEL;cigar|2fb4442e-82ce-441e-99f5-967e803d35fc] Chromosome_3 RepeatMasker similarity 13650932 13651308 3.7 + . ID=Motif:rnd-5_family-3824..1769-2162#83910 375 Chromosome_4 3724167 3725709 DEL;1;None;None 1.0 [Chromosome_4|3724167|3725709|DEL;cigar|04e14531-dd48-4da3-8386-8fa5c0d31a88] Chromosome_4 RepeatMasker similarity 3724168 3724558 3.4 - . ID=Motif:rnd-5_family-3824..1769-2167#108415 391 Chromosome_4 3724167 3725709 DEL;1;None;None 1.0 [Chromosome_4|3724167|3725709|DEL;cigar|04e14531-dd48-4da3-8386-8fa5c0d31a88] Chromosome_4 RepeatMasker similarity 3725358 3725707 0.9 - . ID=Motif:rnd-5_family-3824..618-962#108425 350 Chromosome_8 3875115 3876378 DEL;1;None;None 1.0 [Chromosome_8|3875115|3876378|DEL;cigar|bc18a171-d6da-4e52-b895-55f20b0f603d] Chromosome_8 RepeatMasker similarity 3875527 3875878 22.2 + . ID=Motif:rnd-5_family-3824..1109-1432#43663 352 Chromosome_8 5192359 5193624 DEL;1;None;None 1.0 [Chromosome_8|5192359|5193624|DEL;cigar|3b657f3f-05e2-49fd-9c3d-d27ae0408b84] Chromosome_8 RepeatMasker similarity 5192370 5192776 3.3 - . ID=Motif:rnd-5_family-3824..1769-2162#46002 407 Chromosome_8 5192359 5193624 DEL;1;None;None 1.0 [Chromosome_8|5192359|5193624|DEL;cigar|3b657f3f-05e2-49fd-9c3d-d27ae0408b84] Chromosome_8 RepeatMasker similarity 5193276 5193634 0.6 - . ID=Motif:rnd-5_family-3824..618-962#46012 349
Explore transpositions visually
Create sorted Tabix indices of each bed file.
1
ml tabix; for f in *bed; do sort -k1,1V -k2,3n $f >${f%.*}sorted.bed;bgzip ${f%.*}sorted.bed; tabix -p bed ${f%.*}sorted.bed.gz; done
Viewing your best candidates visually is probably the best way to determine if your TE jump is real. There are a few options to do this. If your genome is hosted online in JBrowse. Go there, select the appropriate genome from “Genome”, then select “Track” and “open track file or URL”. There you should select files and upload your bed.gz files and your .tbi indices. If you do not have your genome in an online browser, you can still view by downloading JBrowse desktop here or with Integrated Genomics Viewer (https://igv.org/doc/desktop/)
To use JBrowse desktop with your custom genome, you will have to create all of the proper indices. Note: getting the sorting right on gene.gff files can be difficult
- .fai index of genome
- .fasta file of genome
- sorted and indexed gff/bed file of repeats
- the SVIM indexed bed files
- sorted.bam file and sorted.bam.bai index
1
2
3
ml samtools; samtools faidx genome.fa
ml tabix; bgzip RepeatMaskerFormatted.gff3; tabix -p gff3 RepeatMaskerFormatted.gff3.gz
ml tabix; bgzip genes.gff3; tabix -p gff3 genes.gff3.gz
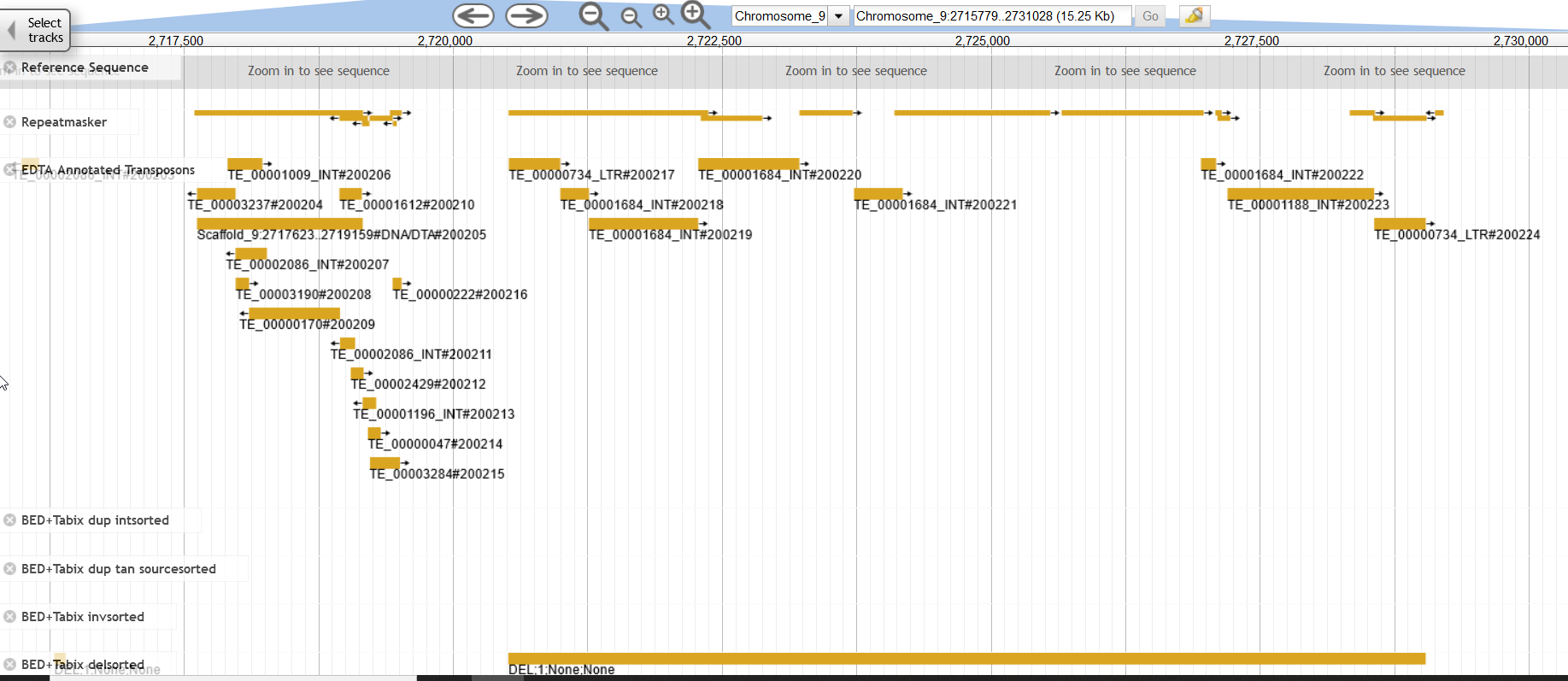
An example of a putative deletion caused by a transposon (excision in the reads/insertion in the genome). Also demonstrating the utility of having multiple repeat annotations for your genome. While my repeat annotations were generic for Repeatmasker/modeler, EDTA provided some intuitive information, that this deletion is bordered by LTR elements, a retrotransposon.
1
Chromosome_9 2720515 2729040 DEL;1;None;None 1.0 [Chromosome_9|2720515|2729040|DEL;cigar|68340730-b565-45db-9f86-4d2bc128e721]
Conclusion
By following this workflow, you can effectively analyze structural variation in your genome and identify actively jumping transposons. This approach combines SVIM’s powerful variant detection with repeat annotation data to highlight regions of potential TE activity, providing valuable insights into genome evolution and modification.
References
- https://github.com/lh3/minimap2
- https://github.com/eldariont/svim
- https://jbrowse.org/blog/