
Introduction to Sequence Read Archive
Sequence Read Archive (SRA) is a publicly accessible repository that serves as a centralized resource for storing and sharing high-throughput sequencing data. It is maintained by the National Center for Biotechnology Information (NCBI), which is a part of the United States National Library of Medicine (NLM).
The SRA database: https://www.ncbi.nlm.nih.gov/sra primarily focuses on storing next-generation sequencing (NGS) data generated from a variety of platforms, such as Illumina, Ion Torrent, PacBio, and others. These sequencing technologies generate vast amounts of raw sequence data, often referred to as reads which can be used for diverse biological and biomedical research purposes.
The SRA database provides a comprehensive infrastructure to organize, store, and retrieve sequencing data. It supports data compression techniques to optimize storage space while maintaining data integrity. The stored data is annotated with essential metadata, including:
- experimental details,
- sample information,
- sequencing platforms, and
- library preparation methods.
Once the desired datasets are identified, users can download the raw sequencing data for further analysis using their preferred bioinformatics tools and pipelines.
Retrieve data from SRA
Researchers and institutions can submit their raw sequencing data to the SRA database, making it available for the scientific community. It enables scientists to access a vast collection of sequencing data from a wide range of organisms and research areas, promoting data reuse, meta-analyses, and the discovery of new insights.
Users can access the SRA database through the NCBI website or programmatically via application programming interfaces (APIs). Both approaches offers powerful search functionalities, allowing users to find specific datasets based on keywords, accession numbers, study details, organism information, or experimental attributes.
Search for data online
The SRA database offers flexible search capabilities, allowing users to search by:
- keywords
- data accession identifiers
- advanced search builder
This ensures that users can efficiently find the datasets they need for their research.
KEYWORDS
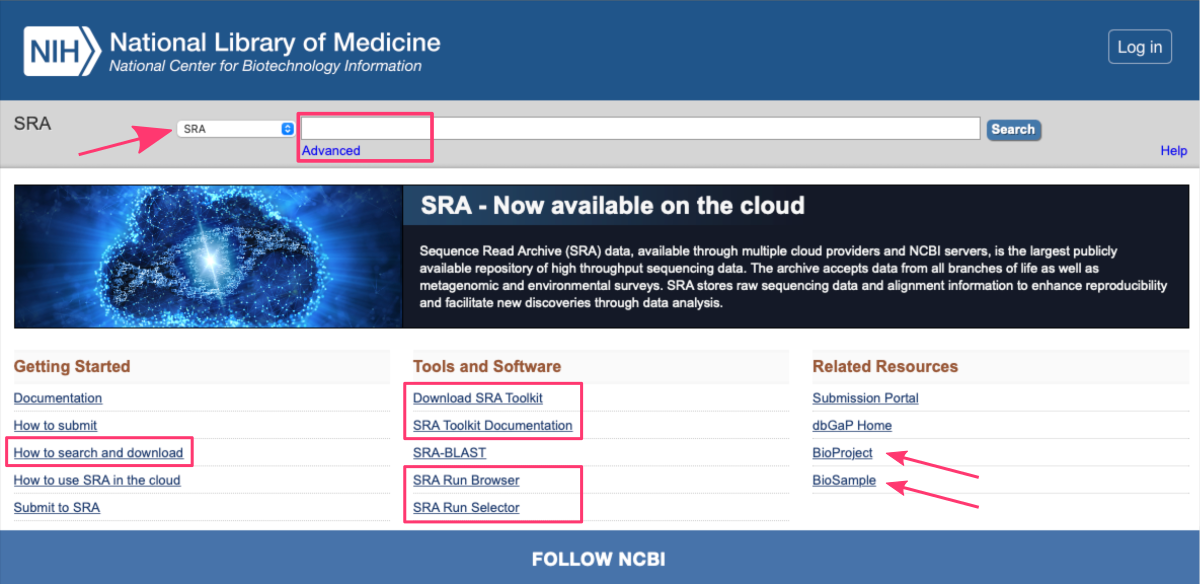
Users can enter keywords related to their research interest in the search box on the SRA database homepage ⤴. These keywords can include gene names, organism names, disease terms, experimental conditions, or any other relevant information. The database will return results that match the provided keywords.
For example, let’s search results for: bumble bee worker

If you want to narrow down your search results, you can utilize various search filters available on the right and left-hand side of the search results page.
DATA ACCESSION IDs
If users already know the specific accession numbers or study names of the datasets they are looking for, they can directly enter those identifiers into the search box. This allows for quick retrieval of the desired datasets without the need for extensive keyword searching.
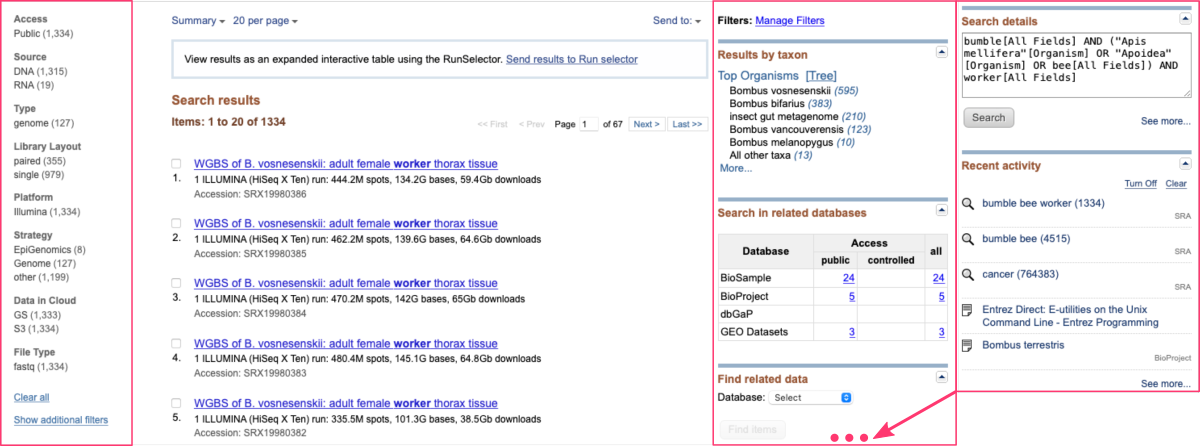
There are several top-level SRA entities and their accessions:
You can also search for BioProjects and GEO Datasets:
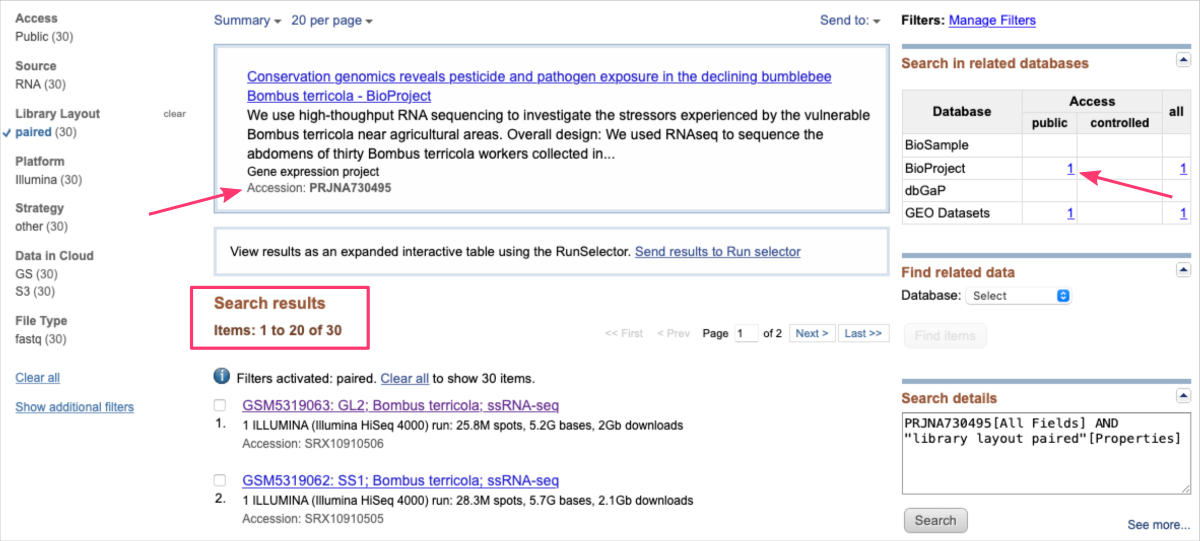
For example, let’s assume you just read the research article PMID: 34181797 ⤴ and have found the Data Availability Statement reffering to BioProject PRJNA730495.

This time, the Search results show all items related to a single BioProject. At the top of the page, you can also find a direct link to the related publication.
EXERCISE FOR YOU:
- Find one specific study: SRP006081 [solution] ⤴
- Find studies with consecutive accessions using wild card: SRP00608* [solution] ⤴
- Find two specific studies: SRP006081 OR SRP006083 [solution] ⤴
ADVANCED SEARCH BUILDER
The SRA database provides an Advanced Search feature that allows users to construct more complex and customized queries. By clicking on the Advanced link below the search box, users can access the Advanced Search Builder. By utilizing this tool, users can create sophisticated queries to retrieve datasets that match their specific requirements.
Advanced Search Builder is especially helpful when users have complex search criteria or need to refine their search based on multiple parameters.
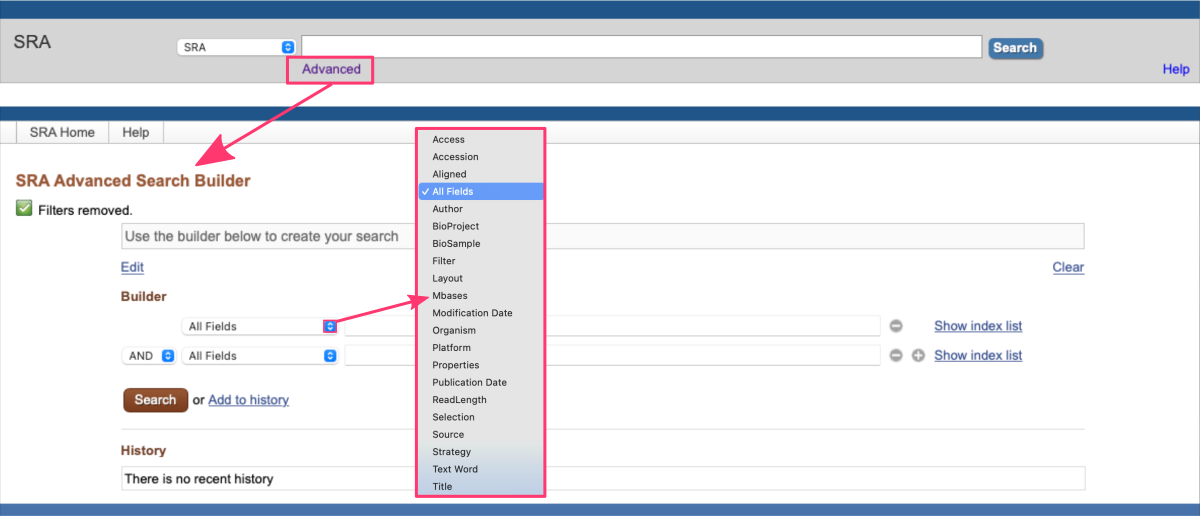
The Advanced Search Builder includes various fields and filters that users can combine to construct their queries. These include:
- Keywords
Users can enter specific keywords related to their research interests. - Filters
Users can apply filters such as organism, platform, study type, library source, instrument model, and more to narrow down the search results. - Operators
Users can use logical operators likeAND,OR, andNOTto combine search terms and create more precise queries.
To get started with Advanced Search Builder please follow the instructions provided in the official SRA docs ⤴.
Obtain run accessions SRR# identifiers
In the SRA database, a Run or Run accession refers to a unique identifier assigned to a specific sequencing run results within a dataset. Each sequencing run corresponds to a set of raw data files generated from a single sequencing library.
Run accessions SRR# are used to download raw sequencing data fom SRA database.
You can obtain run accessions in a few ways:
- A. manually from online SRA
Search results - B. manually from online SRA
Run Selector - C. automatically in the command line
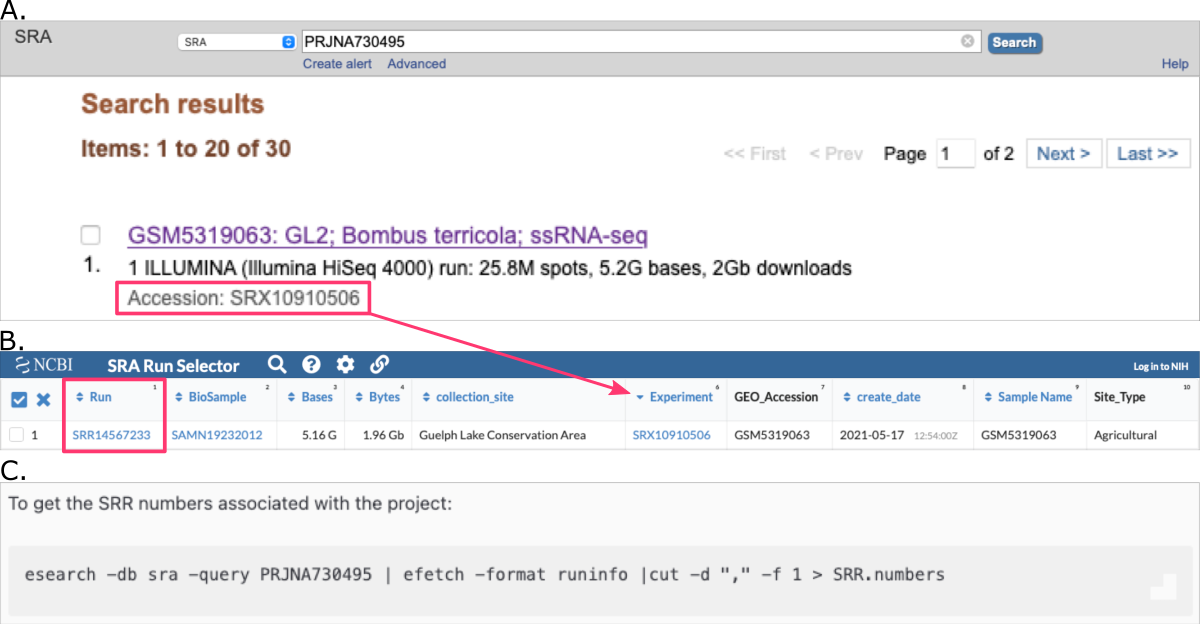
A. manually from SRA Search results
Once you refined your SRA search online (see section Search for data online) and see the list of results, explore search results and get run accessions.
-
Review the
Search resultsand check all the items you want to download.
NOTE: If you do not check any box, by default all results will be prepared for download. -
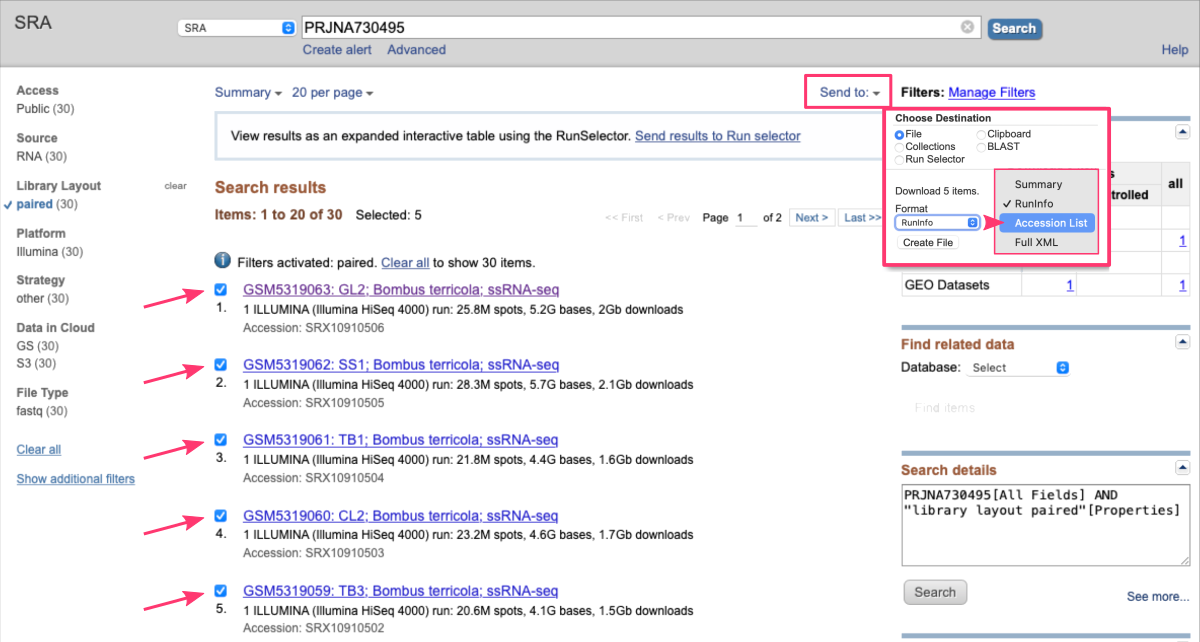
Then, on the top of the page:
• clickSend tobutton and check the radiobuttonFile
• from theFormatdropdown select the optionAccession List
• download this file by clicking theCreate Filebutton
• make sure to move this file to the location where you want to download the raw datasets (i.e., from which you are running the SRA Toolkit)
By default, the downloaded file is named SraAccList.csv and contains a single column with Run accessions:
1
2
3
4
5
6
7
acc
SRR14567204
SRR14567205
SRR14567206
SRR14567207
SRR14567208
...
B. manually from SRA Run Selector
Alternatively, once you refined your SRA search online (see section Search for data online) and see the list of results, you can easily switch to the SRA Run Selector view. This online SRA tool provides a powerful and interactive interface that allows users to filter and sort the sequencing runs based on their preferences.
The Run Selector view enhances the usability of the SRA database by presenting sequencing run information in a structured and interactive manner. It enables users to efficiently explore and select the sequencing runs that meet their specific research requirements, facilitating downstream analysis and data retrieval.
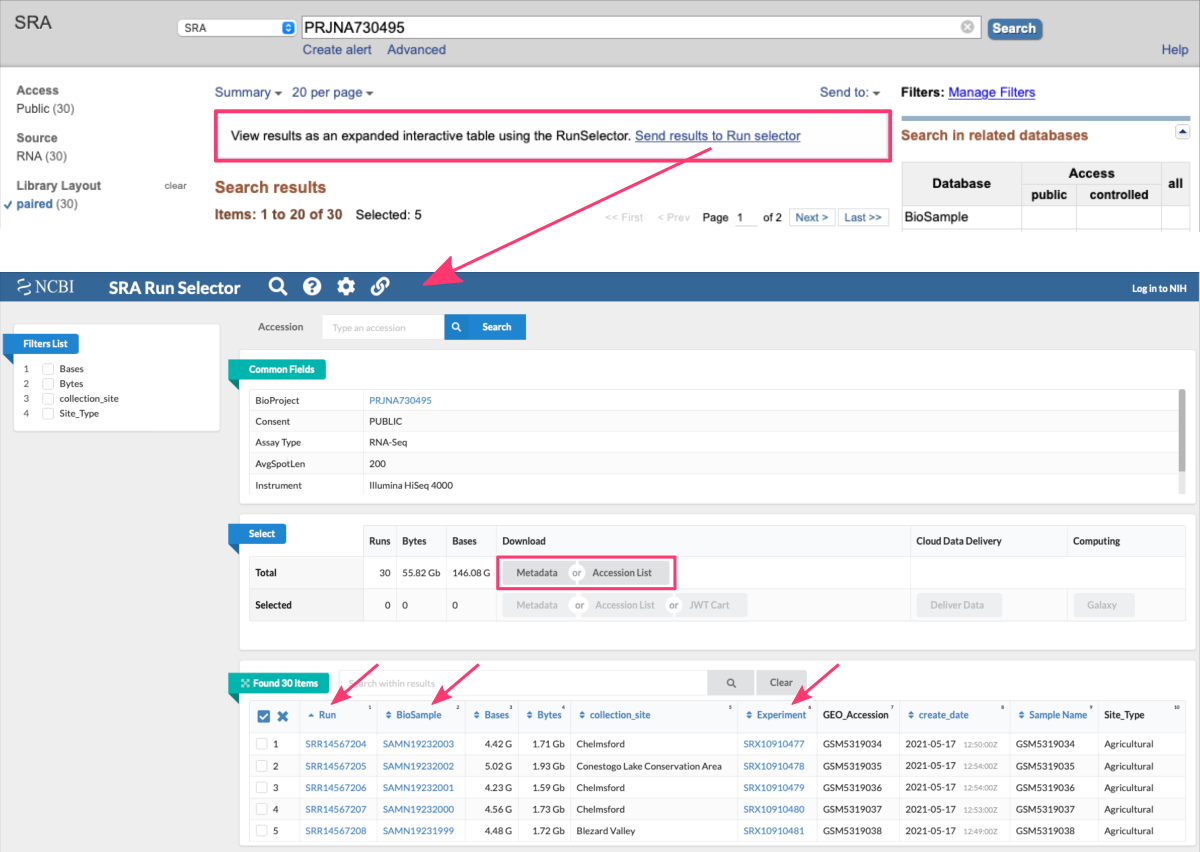
Here are some key features and functionalities of the Run Selector:
-
Filtering
Users can apply filters to refine the list of sequencing runs displayed in the table. Filters are available for various attributes such as organism, library strategy, platform, read length, and more. By selecting specific filter options, users can narrow down the runs to those that match their criteria. -
Sorting
Users can sort the sequencing runs based on specific columns in ascending or descending order. This allows for easy organization and identification of runs based on desired characteristics, such as release date or read length. -
Selection
Users can select one or multiple runs from the table to further explore or download the associated sequencing data. By selecting specific runs, users can proceed to download the data files or access more detailed information about the selected runs. -
Run details
Clicking on a specificRunin the table provides additional details about that particular sequencing run. This may include information such as:- the experiment design,
- library construction protocols,
- experimental factors,
- and other relevant metadata associated with the run.
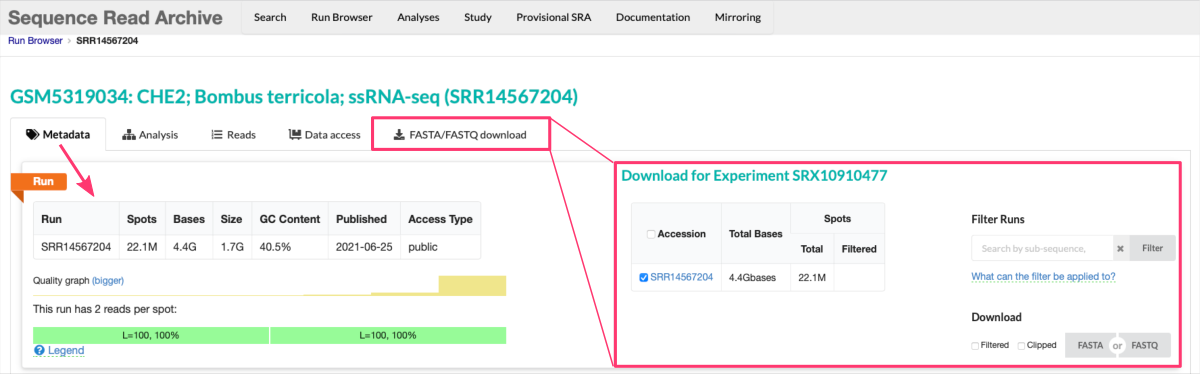
From there, you can also display
Readsand downloadFASTA/FASTQfiles for a given Run accession.
Note, there is a download limit for 5 Gbases of Total Bases. Larger files need to be downloaded via SRA Toolkit.
C. via command line using EDirect tools
To obtain run accessions from the command line you will need the esearch and efetch tools which are included within NCBI Entrez Direct utilities ⤴ (also known as E-utilities).
The esearch command is used to query various NCBI databases, including the SRA database, to retrieve specific information or search results.
The efetch command with the -format runinfo option will retrieve the information for the matching datasets, including the run accessions.
These tools allow you to perform searches and retrieve data programmatically from the command line.
INSTALL EDIRECT (E-utilities)
To use the esearch and efetch commands, you would need to install E-utilities (i.e., the NCBI Entrez Direct utilities, EDirect), which can be obtained from the NCBI website. You can find more information and download instructions for the EDirect at this link: https://www.ncbi.nlm.nih.gov/books/NBK179288/#chapter6_Getting_Started
RETRIEVE SRRs
Once you have installed NCBI’s EDirect, you can use the esearch command to search the SRA database and obtain the necessary information, such as Run accessions, using the efetch command.
The general syntax is as follows:
1
esearch -db sra -query "your search terms" | efetch -format runinfo
Replace “your search terms” with the relevant keywords or search criteria.
For example, to get the SRR numbers associated with the BioProject PRJNA730495:
1
esearch -db sra -query PRJNA730495 | efetch -format runinfo > runinfo.csv
The output file runinfo.csv will contain a table with various details, including the Run accessions (in the first column).
1
2
3
4
5
6
Run,ReleaseDate,LoadDate,spots,bases,spots_with_mates,avgLength,size_MB,AssemblyName,download_path,Experiment,LibraryName,LibraryStrategy,LibrarySelection,LibrarySource,LibraryLayout,InsertSize,InsertDev,Platform,Model,SRAStudy,BioProject,Study_Pubmed_id,ProjectID,Sample,BioSample,SampleType,TaxID,ScientificName,SampleName,g1k_pop_code,source,g1k_analysis_group,Subject_ID,Sex,Disease,Tumor,Affection_Status,Analyte_Type,Histological_Type,Body_Site,CenterName,Submission,dbgap_study_accession,Consent,RunHash,ReadHash
SRR14567204,2021-06-25 18:25:06,2021-05-17 12:50:50,22110877,4422175400,22110877,200,1750,,https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos3/sra-pub-zq-24/SRR014/14567/SRR14567204/SRR14567204.lite.1,SRX10910477,,RNA-Seq,cDNA,TRANSCRIPTOMIC,PAIRED,0,0,ILLUMINA,Illumina HiSeq 4000,SRP320091,PRJNA730495,3,730495,SRS9000934,SAMN19232003,simple,207648,Bombus terricola,GSM5319034,,,,,,,no,,,,,GEO,SRA1233078,,public,EEDE9E10B2038FD62970B54B892532EE,C9297792C29DD79798BDE466149A11C3
SRR14567205,2021-06-25 18:25:06,2021-05-17 12:54:45,25106715,5021343000,25106715,200,1975,,https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos3/sra-pub-zq-24/SRR014/14567/SRR14567205/SRR14567205.lite.1,SRX10910478,,RNA-Seq,cDNA,TRANSCRIPTOMIC,PAIRED,0,0,ILLUMINA,Illumina HiSeq 4000,SRP320091,PRJNA730495,3,730495,SRS9000933,SAMN19232002,simple,207648,Bombus terricola,GSM5319035,,,,,,,no,,,,,GEO,SRA1233078,,public,179BB8F87C8E7AEA09AFD31EA47B17CB,15F09D8C96B02C734C1FD0701A5BACF9
SRR14567206,2021-06-25 18:25:06,2021-05-17 12:54:14,21165129,4233025800,21165129,200,1624,,https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos3/sra-pub-zq-24/SRR014/14567/SRR14567206/SRR14567206.lite.1,SRX10910479,,RNA-Seq,cDNA,TRANSCRIPTOMIC,PAIRED,0,0,ILLUMINA,Illumina HiSeq 4000,SRP320091,PRJNA730495,3,730495,SRS9000935,SAMN19232001,simple,207648,Bombus terricola,GSM5319036,,,,,,,no,,,,,GEO,SRA1233078,,public,F59A9C00EED65826E0F40DF85CA0E512,F76970E0700A354530E7C0F28B0E0599
SRR14567207,2021-06-25 18:25:06,2021-05-17 12:53:18,22777492,4555498400,22777492,200,1775,,https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos3/sra-pub-zq-24/SRR014/14567/SRR14567207/SRR14567207.lite.1,SRX10910480,,RNA-Seq,cDNA,TRANSCRIPTOMIC,PAIRED,0,0,ILLUMINA,Illumina HiSeq 4000,SRP320091,PRJNA730495,3,730495,SRS9000936,SAMN19232000,simple,207648,Bombus terricola,GSM5319037,,,,,,,no,,,,,GEO,SRA1233078,,public,9A865DAAC8CB2F9336A553C44D371CCB,509CFAFFD98F8877B09D19E31305F0EE
...
You can modify the above command to retrieve only the SRR# identifiers:
1
esearch -db sra -query PRJNA730495 | efetch -format runinfo | cut -d "," -f 1 > SRR.numbers
The content of the SRR.numbers file will be like that:
1
2
3
4
5
6
Run
SRR14567204
SRR14567205
SRR14567206
SRR14567207
...
Download SRA data files
To download raw data files from SRA database you have a few options. All of them assume that you have a list of Run accessions, i.e. the SRR# identifiers for data gathered for samples of your interest.
Please see section Obtain run accessions SRR# to learn how to retrieve identifiers of samples from SRA database.
Once you have a file with all Run accessions SRR#, for example:
1
2
3
4
SRR14567204
SRR14567205
SRR14567206
SRR14567207
please follow the instructions for the approach of your choice:
- command line approaches:
A. using SRA Toolkit
The NCBI’s SRA Toolkit provides command-line utilities to download and manipulate SRA data. This software suite has been configured to connect to NCBI databases (including SRA) and download data via FTP or HTTPS.
Install SRA Toolkit on your local machine
Visit the NCBI SRA Toolkit repository (https://github.com/ncbi/sra-tools/wiki/01.-Downloading-SRA-Toolkit) to download and install the SRA Toolkit appropriate for your operating system.
Load SRA Toolkit module on the HPC infrastructure
On an HPC (High-Performance Computing) cluster, it is common for administrators to preinstall various software packages and tools to provide users with a consistent and accessible computing environment. The SRA Toolkit, including its command-line utilities, is one such tool that may be preinstalled on the HPC cluster.
When the SRA Toolkit is installed on an HPC cluster, it is typically made available as a module. A module is a software management system that allows users to load or unload specific software packages and their associated environments.
- Check available modules:
1
module avail sratoolkit
This command displays the software variants that have been preinstalled and are ready to be loaded.
For example:sratoolkit/3.0.0-6ikpzzj - Load the selected SRA Toolkit module:
1
module load sratoolkit/3.0.0-6ikpzzj
Get started with SRA Toolkit tools
Once the module is loaded, you can use tools available in the SRA Toolkit to download the raw Sequence Reads from the SRA database.
The SRA Toolkit provides a set of command-line utilities that facilitate the retrieval, manipulation, and analysis of data from the NCBI’s Sequence Read Archive (SRA) database.
Use the name of the command-line utility followed by the -h flag to learn more about the options available for each tool.
sratoolkit command |
description |
|---|---|
| prefetch | Downloads binary SRA data files based on accession numbers or a text file containing accession numbers. It retrieves the data files and saves them locally for further processing. |
| fastq-dump | Converts SRA data files into FASTQ format, which is a common file format for storing sequence data. It extracts the raw sequence reads and quality scores from the SRA files. |
| sam-dump | Converts SRA data files into SAM/BAM format, which is used for storing sequence alignment information. It allows you to extract alignment data from SRA files in a readable format. |
| vdb-validate | Performs validation checks on SRA data files to ensure their integrity and adherence to formatting standards. It can be used to verify the integrity of downloaded SRA files. |
| vdb-config | Modifies configuration settings for the SRA Toolkit, such as specifying the location of downloaded files or adjusting caching options. |
| fasterq-dump | A faster alternative to fastq-dump, specifically designed for downloading and converting SRA data files into FASTQ format. It provides improved performance for large datasets. |
The toolkit offers additional utilities and options for more advanced operations, such as filtering, subsetting, and quality control of SRA data.
The simple command to fetch a SRA file in FASTQ format, you can use the fastq-dump command:
1
fastq-dump SRR447882
This will download the SRA file (in sra format) and then convert them to fastq file for you.
If your SRA file is paired, you will still end up with a single fastq file, since, fastq-dump, by default writes them as interleaved file. To change this, you can provide --split-files argument.
1
fastq-dump --split-files SRR447882
The downloaded fastq files will have @SRR number suffixed on all header lines of the file:
1
2
3
4
5
6
7
8
9
10
11
12
13
head -n 12 SRR447882_1.fastq
@SRR447882.1.1 HWI-EAS313_0001:7:1:6:844 length=84
ATTGATCATCGACCAGAGNCTCATACACCTCACCCCACATATGTTTCCTTGCCATAGATCACATTCTTGNNNNNNNGGTGGANA
+SRR447882.1.1 HWI-EAS313_0001:7:1:6:844 length=84
BBBBBB;BB?;>7;?<?B#AA3@CBAA?@BAA@)=6ABBBBB?ACA;0A=257?A7+;;&########################
@SRR447882.2.1 HWI-EAS313_0001:7:1:6:730 length=84
AGTTGATTGTGATATAGGNGTCTATCGACATTGATGCATAGGTCCTCTATTAAACTTGTTTTGTGATGTNNNNNNNTTTTTTNA
+SRR447882.2.1 HWI-EAS313_0001:7:1:6:730 length=84
A?@B:@CA:=?BCBC:2C#7>BACB??@4@B@<=>;'>@>3:86>=6@=B@B<;)@@###########################
@SRR447882.3.1 HWI-EAS313_0001:7:1:6:1343 length=84
CATCAATGCAAGGATTGTNCCATTGGTAACAATTCCACTCCTAACTTGTCAATTGATTTTCATATAACTNNNNNNNCCAAAANT
+SRR447882.3.1 HWI-EAS313_0001:7:1:6:1343 length=84
BCB@BBC+5BCA>BABBA#@4BCCA>?CBBB4CB(*ABB?ABBAACCB8ABBB?(<<B?:########################
Although, this normally does not affect any programs, some programs might throw an error saying that it can’t process these fastq files. To avoid this, you an request the file to be in the orignal format (--origfmt). Also, note that if you’re downloading files in bulk, you can save a lot of space by compressing them in gzip format (--gzip).
1
fastq-dump --split-files --origfmt --gzip SRR447882
The fastq-dump is also capable of doing:
- Additional filtering or clipping of the downloaded reads: to remove reads with poor quality or to trim adapters. Although, this will work for the single end reads, for paired-end reads it may cause differential treatment for each pairs and might not be usable for mapping programs that needs strict pairs.
- Compressed format: either as gzipped or bzipped files using
--gzipor--bzip2options. - fasta format: by using the
--fastaoption
B. using Linux commands
The NCBI Team, on October 17, 2019, announced the decommission of the SRA Fuse/FTP site and encouraged the users to try the SRA Toolkit as the recommended method of downloading the data from SRA. [source ⤴]
In relation to the above warning, we encourage you to choose an approach based on the use of the SRA Toolkit. See section A. using SRA Toolkit.
In cases were you cannot run the SRA toolkit or any other programs to download the file, you can still try to use the built-in commands of Linux such as wget and curl.
Note that downloading files from the SRA database using this approach is very slow (~1MB/s). Also, the files larger than 5 Gbases are not available to download using this method.
The standard web link for downloading the SRA files is:
https://trace.ncbi.nlm.nih.gov/Traces/sra-reads-be/fastq?acc=SRRNNNNNN
In the past the working link was also this one:
http://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?cmd=dload&run_list=SRRNNNNNN&format=fastq
You need to replace the SRRNNNNNN with the actual SRR number for it to work.
You can either use wget command:
1
wget https://trace.ncbi.nlm.nih.gov/Traces/sra-reads-be/fastq?acc=SRR14567204
or curl command:
1
curl -O https://trace.ncbi.nlm.nih.gov/Traces/sra-reads-be/fastq?acc=SRR14567204
If you have a large list of ids, you can simply loop it over using a while loop
1
2
3
while read line; do
wget https://trace.ncbi.nlm.nih.gov/Traces/sra-reads-be/fastq?acc=${line};
done < list_of_ids
The datasets can also be downloaded from DDBJ or EMBL using the FTP links, but the transfer speeds might be affected if you're not near their servers.
C. using Aspera Connect
Aspera uses high-speed file transfer to rapidly transfer large files and data sets over an existing WAN infrastructure.
To get the SRA files:
1
prefetch --max-size 100G --transport ascp --ascp-path "/path/to/aspera/3.6.2/bin/ascp|/path/to/aspera/3.6.2/etc/asperaweb_id_dsa.openssh" SRRNNNNNN
This usually prefetches the SRA file to your home directory in folder named ncbi. If your home directory does not contain enough space to store all data, you may want to create another directory and softlink to the home. To do this:
1
2
3
cd ~
mkdir -p /project/storage/your_dir/ncbi
ln -s /project/storage/your_dir/ncbi
when you run this, you will have a directory named ncbi in your home, but the data is actually stored in /project/storage/your_dir/ncbi
1
2
3
4
5
ncbi
└── public
└── sra
├── SRR006189.sra
└── SRR006190.sra
Then you can convert the SRA files back to fastq format using fastq-dump command.
1
2
3
for sra file in ~/ncbi/public/sra/*; do
fastq-dump --split-files --origfmt --gzip ${sra};
done
ready-made examples
Download all SRA files related to a BioProject
NCBI Sequence Read Archive (SRA) stores sequence and quality data (fastq files) in aligned or unaligned formats from NextGen sequencing platforms. A BioProject is a collection of biological data related to a single initiative, originating from a single organization or from a consortium. A BioProject record provides users a single place to find links to the diverse data types generated for that project. Often times, once single BioProject will hold a considerable number of experiments and it gets tedious to download them all individually.
Here is the guide to show how to do this in a effecient way:
First, we need the esearch program for Entrez searching. So, navigate to the home directory and run the commands:
1
2
sh -c "$(curl -fsSL https://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/install-edirect.sh)"
sh -c "$(wget -q https://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/install-edirect.sh -O -)"
This will download a number of scripts and several precompiled programs into an “edirect” folder in the user’s home directory. Once you add it to your $PATH, the esearch command will be available for you from any location on the file system.
If you are working on the HPC cluster, make sure to encapsulate the following commands in the script for workload manager (typically SLURM or PBS) and submit the job to the queue. Alternatively, you can start the interactive session on the computing node:
salloc -N 1 -n 4 -t 01:00:00qsub -IIn the next step, load the modules that are needed:
1
2
module load sratoolkit
module load parallel
To get the SRR numbers associated with the project:
1
esearch -db sra -query PRJNA730495 | efetch -format runinfo |cut -d "," -f 1 > SRR.numbers
To download the corresponding SRA data all at once in parallel (limit the number to 4 concurrent downloads) use the following command:
1
parallel --jobs 4 "fastq-dump --split-files --origfmt --gzip {}" ::: SRR.numbers
Use ready-made script for the SLURM workload manager
download_raw_samples.sh
1
2
3
4
5
6
7
8
9
10
11
12
13
#!/bin/bash
#SBATCH --nodes=1
#SBATCH --cpus-per-task=8
#SBATCH --time=01:00:00
module load sratoolkit
module load parallel
project='PRJNA730495'
esearch -db sra -query $project | efetch -format runinfo > runinfo.csv
cat runinfo.csv | cut -d "," -f 1 > SRR.numbers
cat SRR.numbers | parallel fastq-dump --split-files --origfmt --gzip -X 1000 {}
Run in the comman-line:
1
sbatch download_raw_samples.sh